

Pharmacological approaches to restore mitochondrial function
- PMID: 23666487
- PMCID: PMC3896945
- DOI: 10.1038/nrd4023
Pharmacological approaches to restore mitochondrial function
Abstract
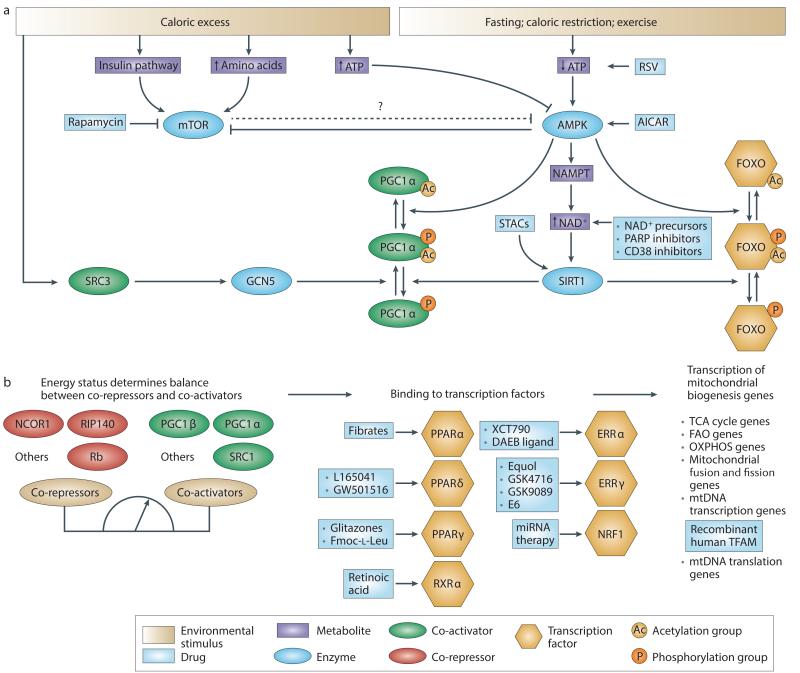
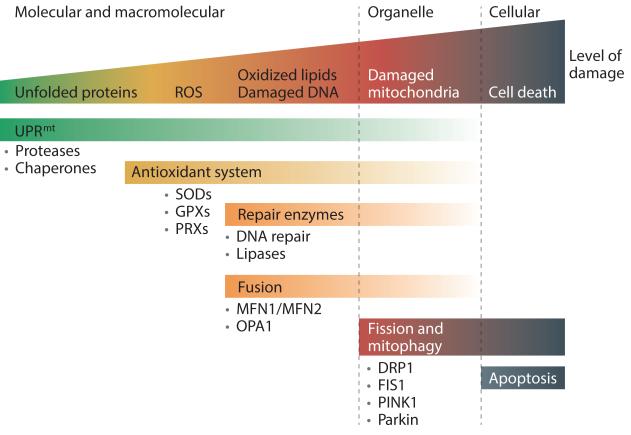
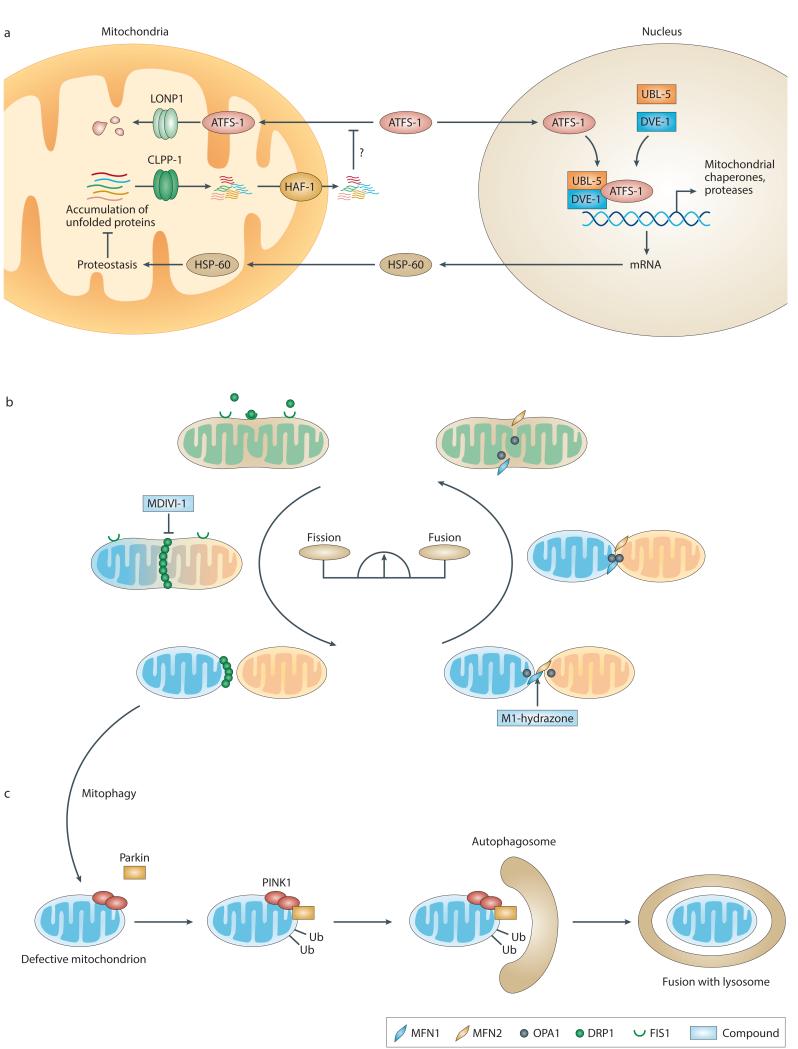
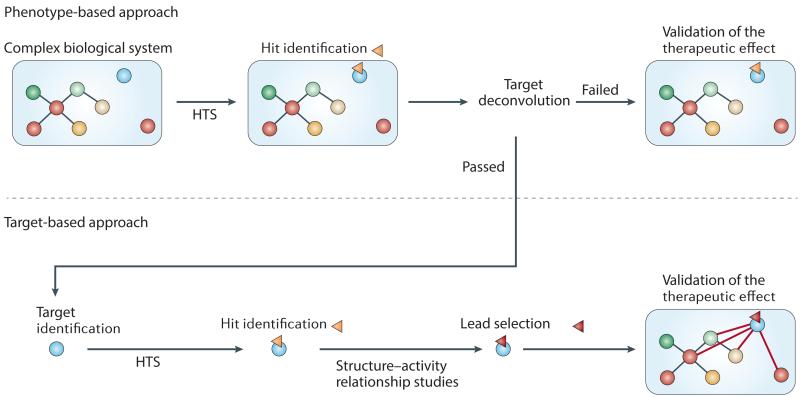
Mitochondrial dysfunction is not only a hallmark of rare inherited mitochondrial disorders but also implicated in age-related diseases, including those that affect the metabolic and nervous system, such as type 2 diabetes and Parkinson's disease. Numerous pathways maintain and/or restore proper mitochondrial function, including mitochondrial biogenesis, mitochondrial dynamics, mitophagy and the mitochondrial unfolded protein response. New and powerful phenotypic assays in cell-based models as well as multicellular organisms have been developed to explore these different aspects of mitochondrial function. Modulating mitochondrial function has therefore emerged as an attractive therapeutic strategy for several diseases, which has spurred active drug discovery efforts in this area.
Figures
References
-
- Wallin IE, Wallin Ivan E. Symbionticism and the origin of species. Williams & Wilkins Company; Baltimore: 1927.
-
- Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–22. - PubMed
-
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–55. - PubMed
-
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–67. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
