

Next-generation sequencing identifies transportin 3 as the causative gene for LGMD1F
- PMID: 23667635
- PMCID: PMC3646821
- DOI: 10.1371/journal.pone.0063536
Next-generation sequencing identifies transportin 3 as the causative gene for LGMD1F
Abstract
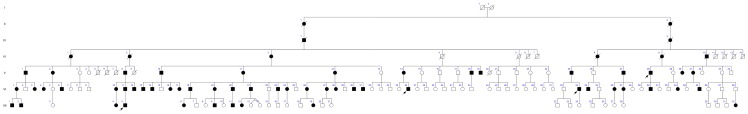
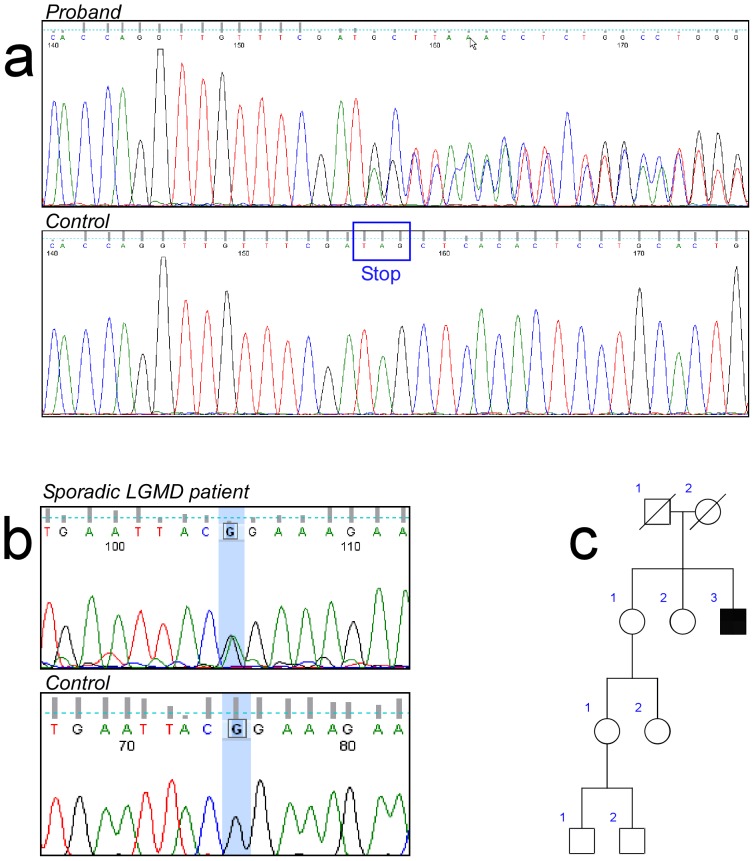
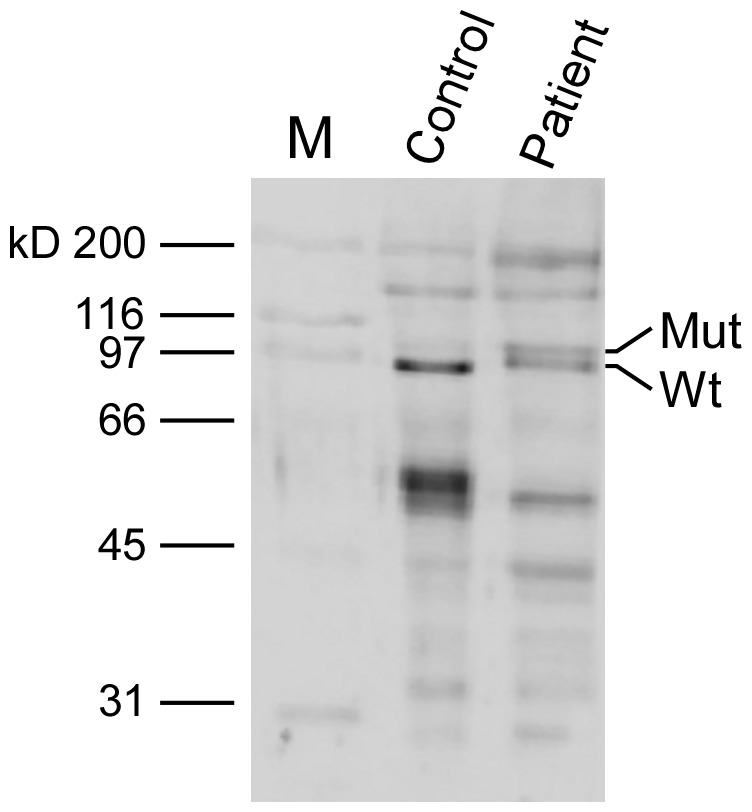
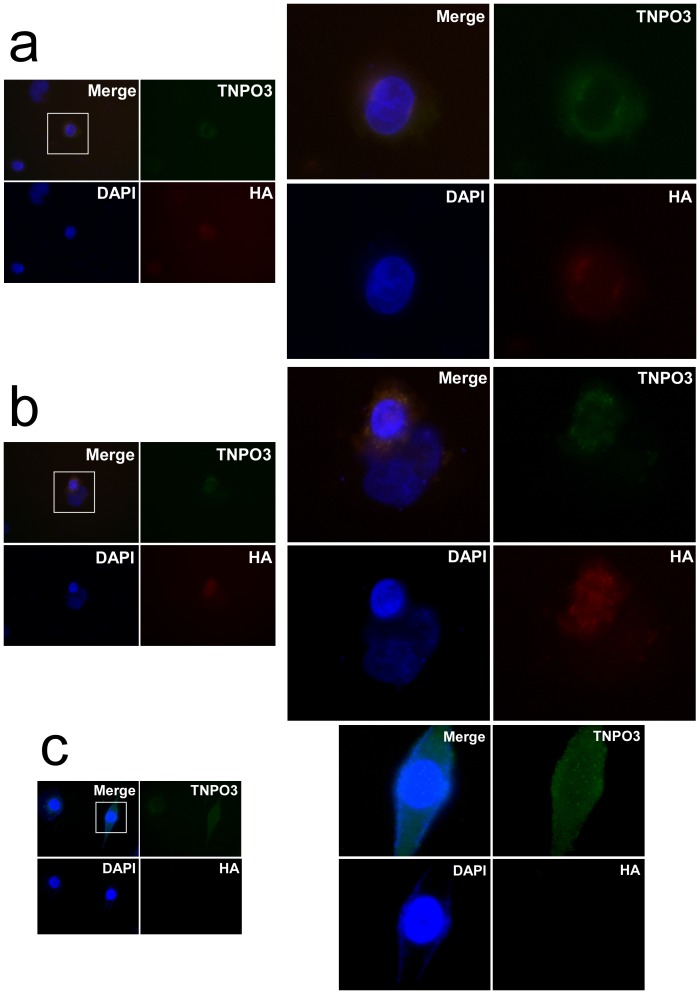
Limb-girdle muscular dystrophies (LGMD) are genetically and clinically heterogeneous conditions. We investigated a large family with autosomal dominant transmission pattern, previously classified as LGMD1F and mapped to chromosome 7q32. Affected members are characterized by muscle weakness affecting earlier the pelvic girdle and the ileopsoas muscles. We sequenced the whole exome of four family members and identified a shared heterozygous frame-shift variant in the Transportin 3 (TNPO3) gene, encoding a member of the importin-β super-family. The TNPO3 gene is mapped within the LGMD1F critical interval and its 923-amino acid human gene product is also expressed in skeletal muscle. In addition, we identified an isolated case of LGMD with a new missense mutation in the same gene. We localized the mutant TNPO3 around the nucleus, but not inside. The involvement of gene related to the nuclear transport suggests a novel disease mechanism leading to muscular dystrophy.
Conflict of interest statement
Figures
References
-
- Nigro V (2003) Molecular bases of autosomal recessive limb-girdle muscular dystrophies. Acta Myol 22: 35–42. - PubMed
-
- Nigro V, Aurino S, Piluso G (2011) Limb girdle muscular dystrophies: update on genetic diagnosis and therapeutic approaches. Curr Opin Neurol 24: 429–436. - PubMed
-
- Fanin M, Nascimbeni AC, Aurino S, Tasca E, Pegoraro E, et al. (2009) Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 72: 1432–1435. - PubMed
-
- Mercuri E, Bushby K, Ricci E, Birchall D, Pane M, et al. (2005) Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscul Disord 15: 164–171. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- TGM11Z06/TI_/Telethon/Italy
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- GUP10006/TI_/Telethon/Italy
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- GUP11006/TI_/Telethon/Italy
- HL-102924/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- GTB12001/TI_/Telethon/Italy
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
