

Computational meta'omics for microbial community studies
- PMID: 23670539
- PMCID: PMC4039370
- DOI: 10.1038/msb.2013.22
Computational meta'omics for microbial community studies
Abstract
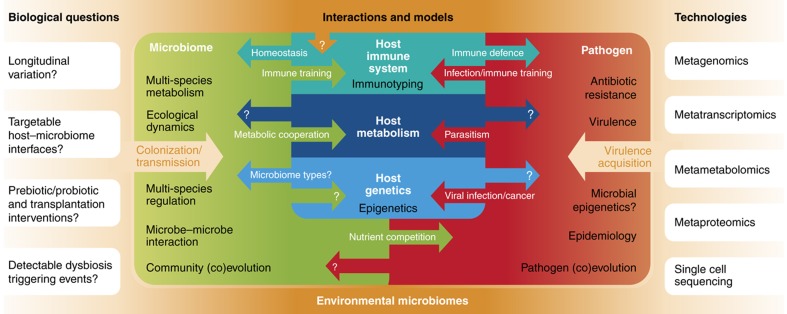
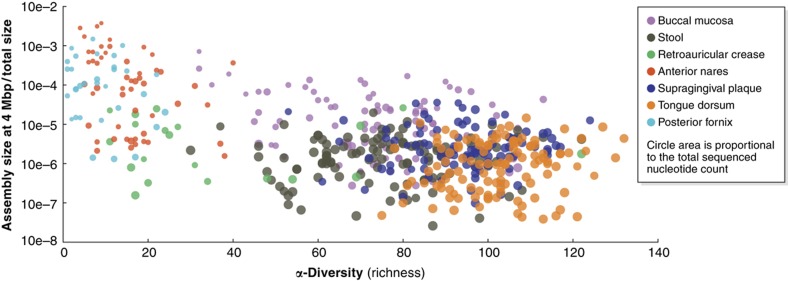
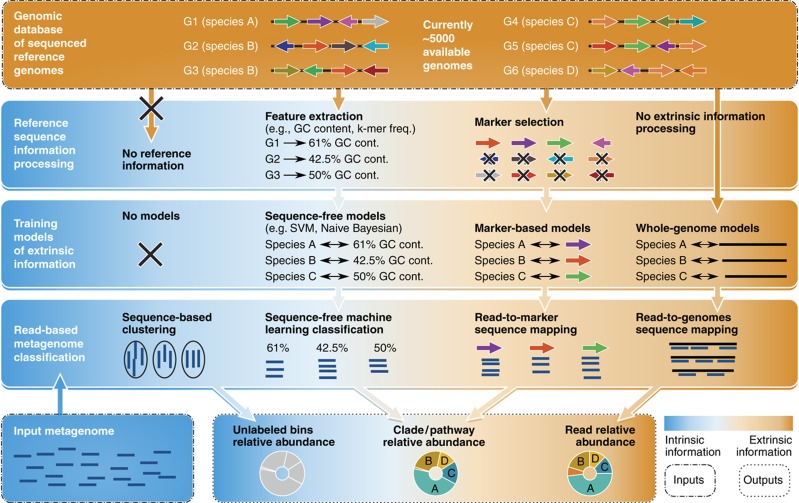
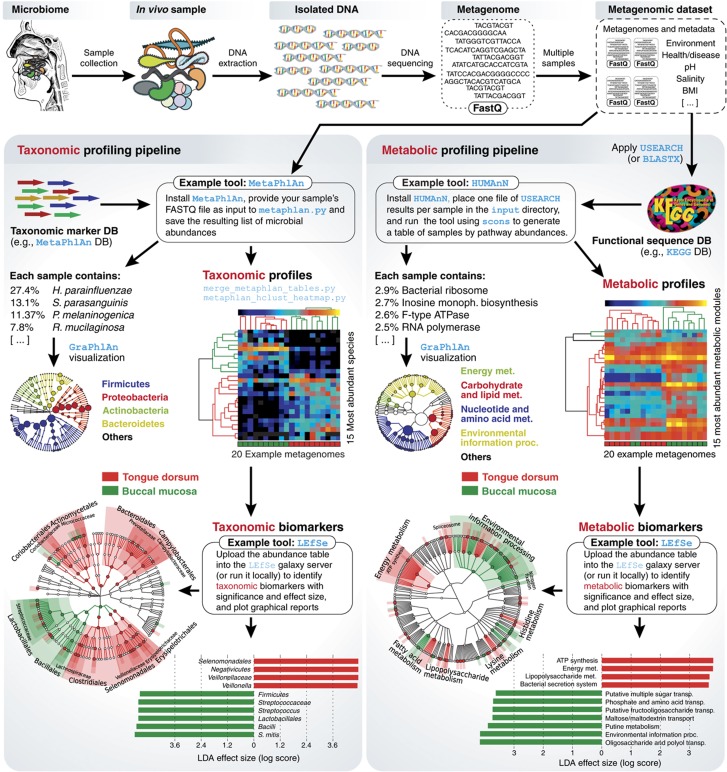
Complex microbial communities are an integral part of the Earth's ecosystem and of our bodies in health and disease. In the last two decades, culture-independent approaches have provided new insights into their structure and function, with the exponentially decreasing cost of high-throughput sequencing resulting in broadly available tools for microbial surveys. However, the field remains far from reaching a technological plateau, as both computational techniques and nucleotide sequencing platforms for microbial genomic and transcriptional content continue to improve. Current microbiome analyses are thus starting to adopt multiple and complementary meta'omic approaches, leading to unprecedented opportunities to comprehensively and accurately characterize microbial communities and their interactions with their environments and hosts. This diversity of available assays, analysis methods, and public data is in turn beginning to enable microbiome-based predictive and modeling tools. We thus review here the technological and computational meta'omics approaches that are already available, those that are under active development, their success in biological discovery, and several outstanding challenges.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methe B, Schloss PD, Gevers D, Mitreva M, Huttenhower C (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8: e1002358. - PMC - PubMed
-
- Arumugam M, Harrington ED, Foerstner KU, Raes J, Bork P (2010) SmashCommunity: a metagenomic annotation and analysis tool. Bioinformatics 26: 2977–2978 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
