

Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties
- PMID: 23673625
- PMCID: PMC3674249
- DOI: 10.1038/ncomms2835
Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties
Abstract
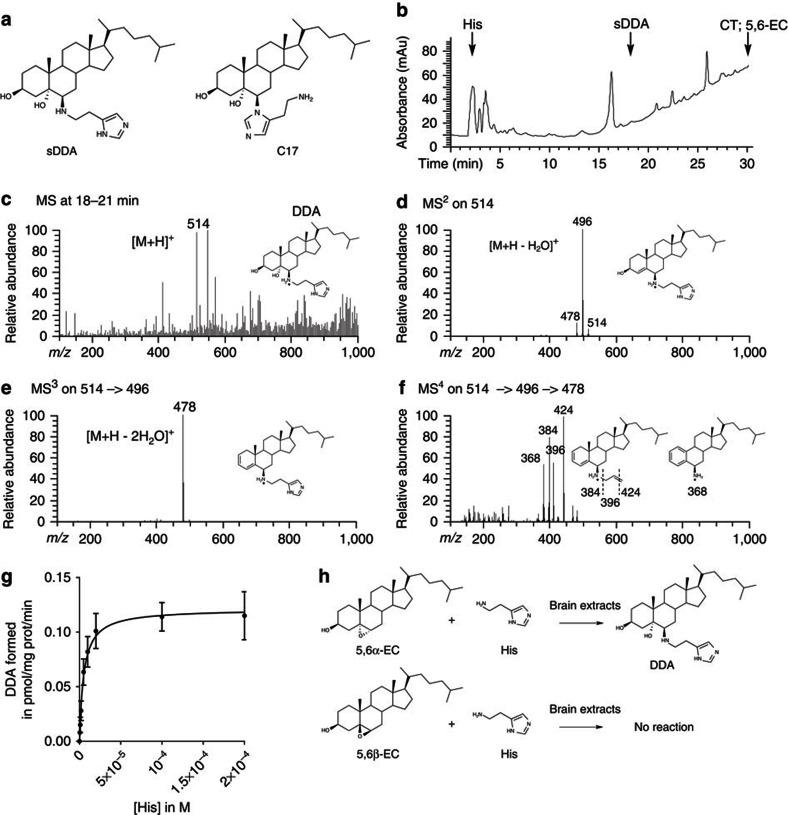
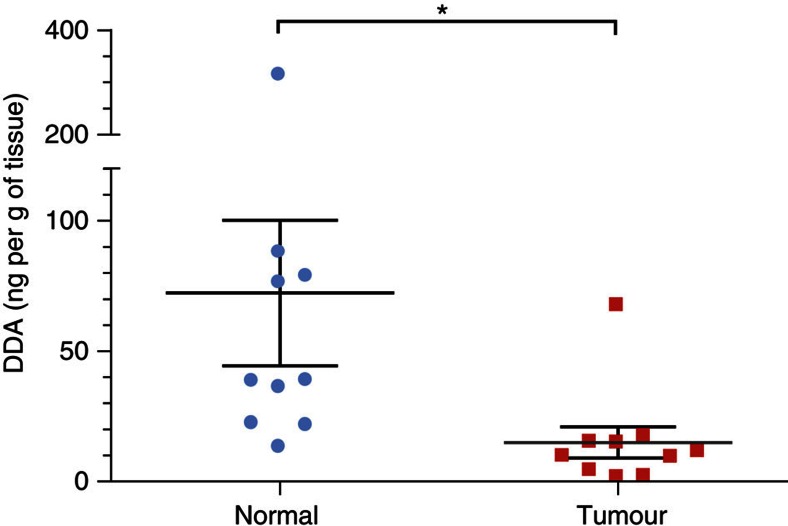
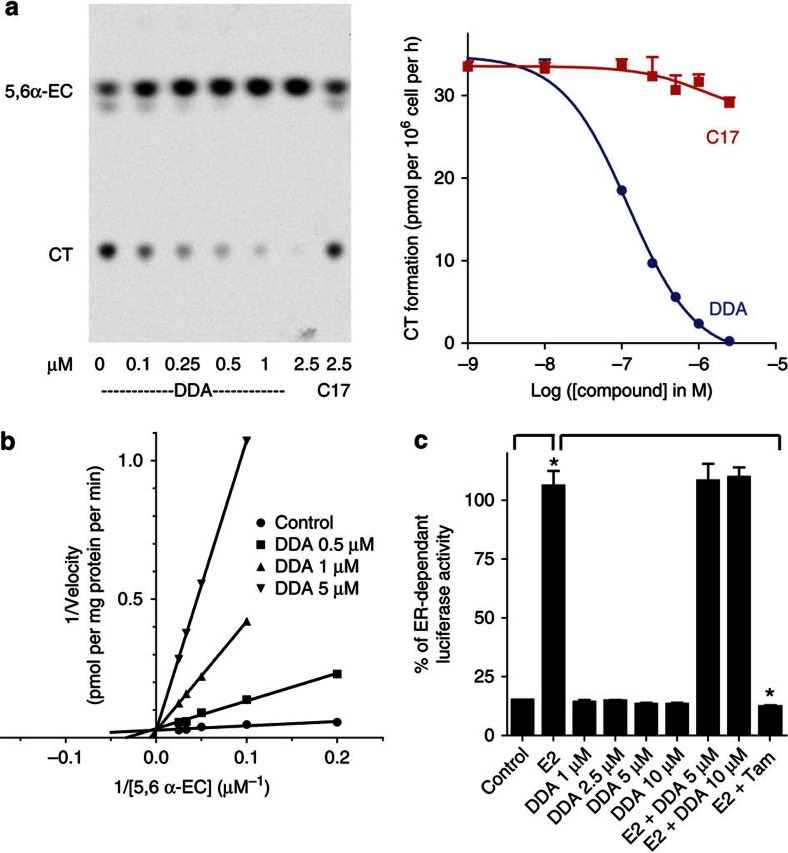
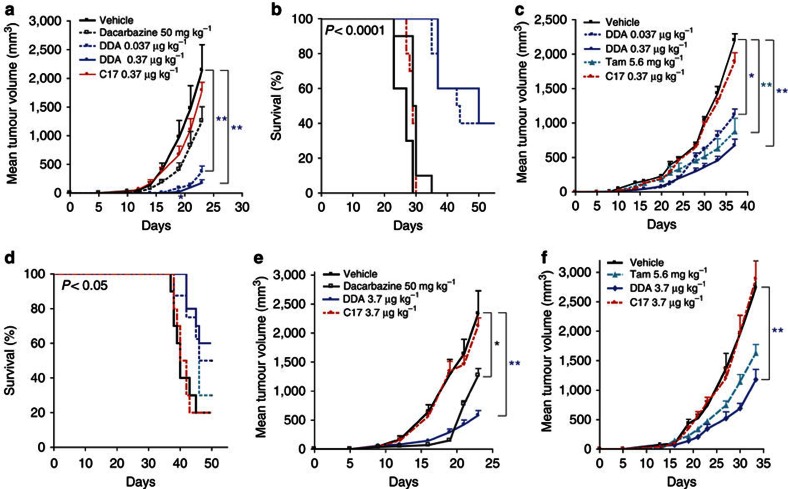
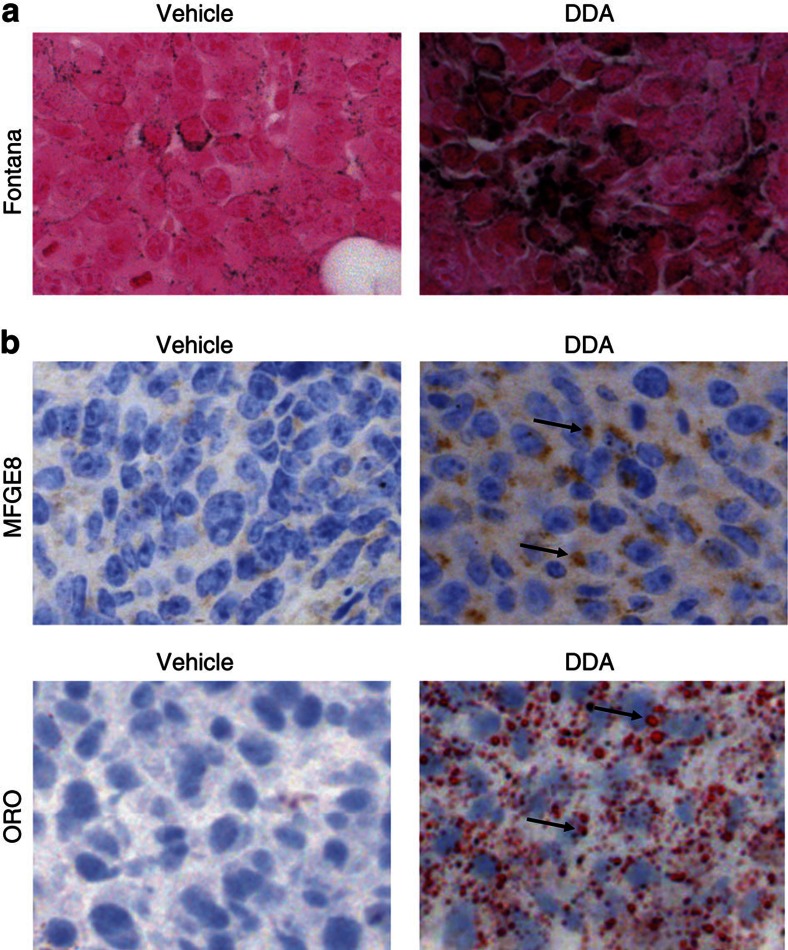
We previously synthesized dendrogenin A and hypothesized that it could be a natural metabolite occurring in mammals. Here we explore this hypothesis and report the discovery of dendrogenin A in mammalian tissues and normal cells as an enzymatic product of the conjugation of 5,6α-epoxy-cholesterol and histamine. Dendrogenin A was not detected in cancer cell lines and was fivefold lower in human breast tumours compared with normal tissues, suggesting a deregulation of dendrogenin A metabolism during carcinogenesis. We established that dendrogenin A is a selective inhibitor of cholesterol epoxide hydrolase and it triggered tumour re-differentiation and growth control in mice and improved animal survival. The properties of dendrogenin A and its decreased level in tumours suggest a physiological function in maintaining cell integrity and differentiation. The discovery of dendrogenin A reveals a new metabolic pathway at the crossroads of cholesterol and histamine metabolism and the existence of steroidal alkaloids in mammals.
Conflict of interest statement
P.D.M., M.R.P. and L.M. are employees of the company Affichem of which S.S.-P. and M.P. are founders. The remaining authors declare no competing financial interests.
Figures
References
-
- Newman J. W., Morisseau C., Hammock B. D. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog. Lipid Res. 44, 1–51 (2005). - PubMed
-
- Poirot M., Silvente-Poirot S., Weichselbaum R. R. Cholesterol metabolism and resistance to tamoxifen. Curr. Opin. Pharmacol. 12, 683–689 (2012). - PubMed
-
- de Medina P. et al.. Importance of cholesterol and oxysterols metabolism in the pharmacology of tamoxifen and other AEBS ligands. Chem. Phys. Lipids 164, 432–437 (2011). - PubMed
-
- Payre B. et al.. Microsomal antiestrogen-binding site ligands induce growth control and differentiation of human breast cancer cells through the modulation of cholesterol metabolism. Mol. Cancer Ther. 7, 3707–3718 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
