

Identifying molecular targets of lifestyle modifications in colon cancer prevention
- PMID: 23675573
- PMCID: PMC3653120
- DOI: 10.3389/fonc.2013.00119
Identifying molecular targets of lifestyle modifications in colon cancer prevention
Abstract
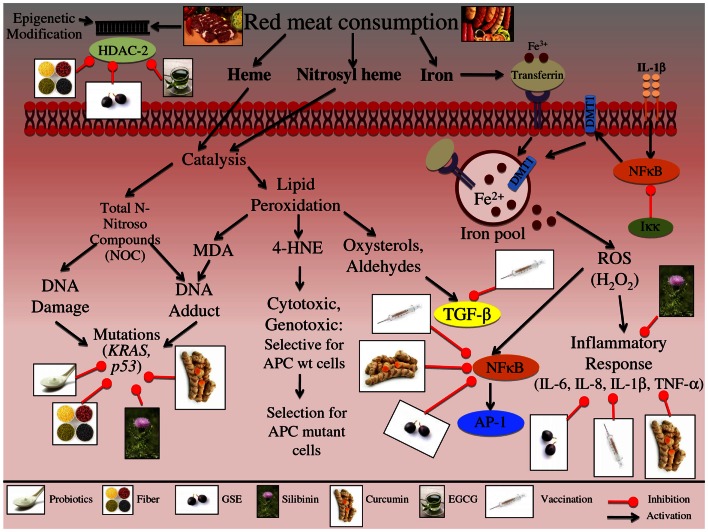
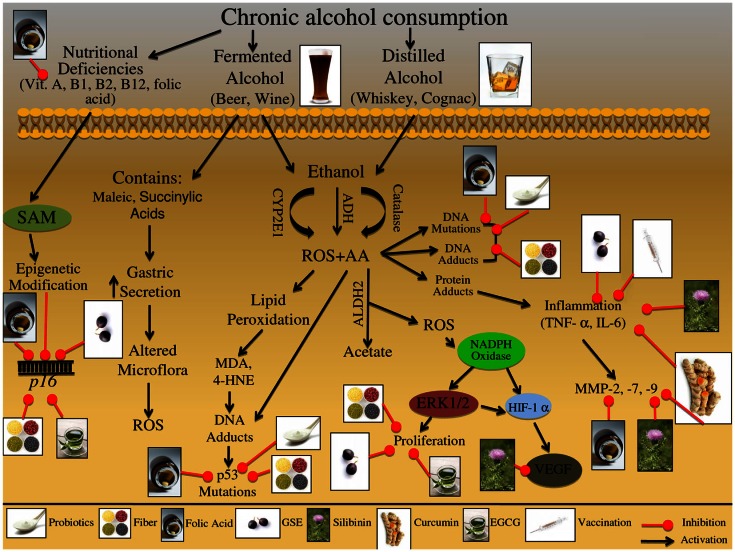
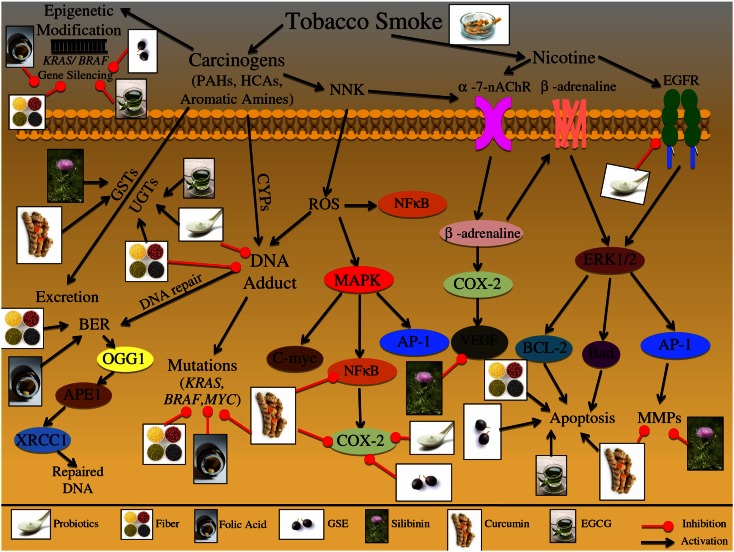
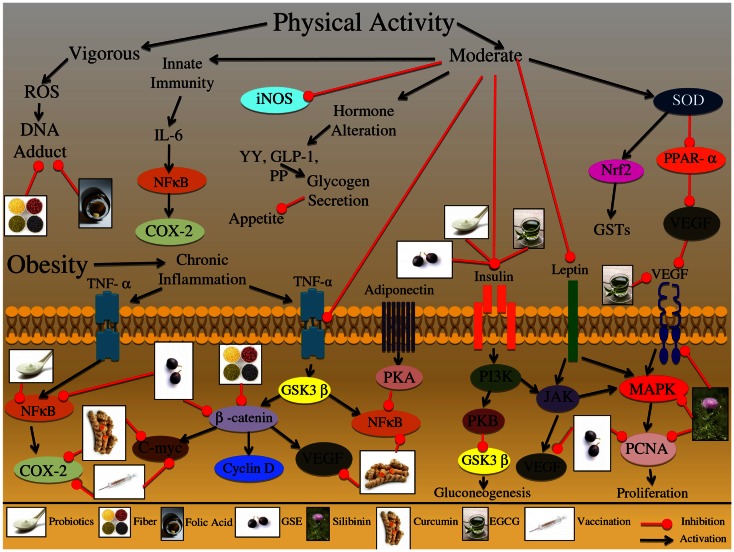
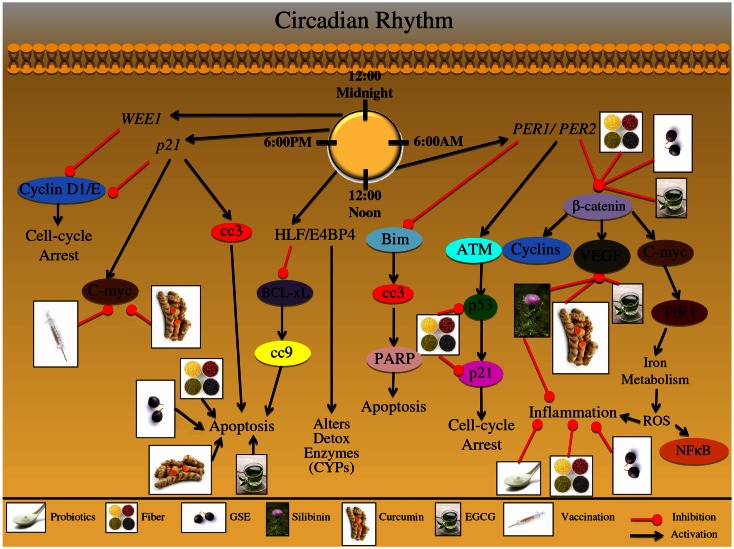
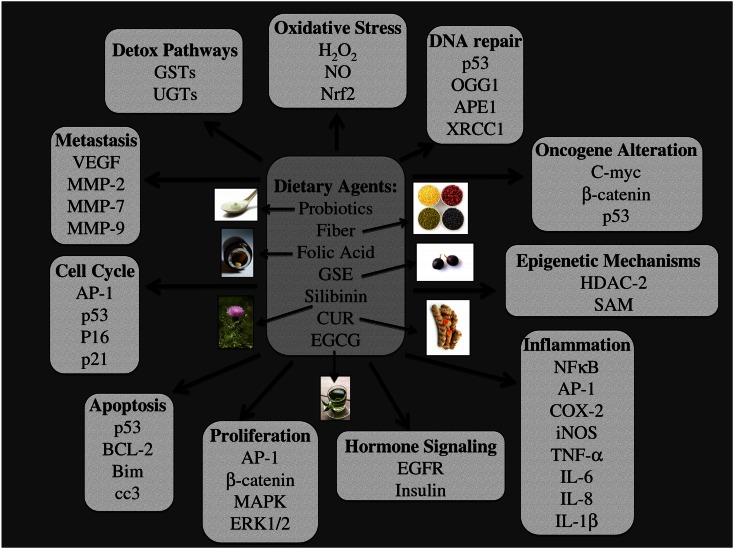
One in four deaths in the United States is cancer-related, and colorectal cancer (CRC) is the second leading cause of cancer-associated deaths. Screening strategies are utilized but have not reduced disease incidence or mortality. In this regard, there is an interest in cancer preventive strategies focusing on lifestyle intervention, where specific etiologic factors involved in cancer initiation, promotion, and progression could be targeted. For example, exposure to dietary carcinogens, such as nitrosamines and polycyclic aromatic hydrocarbons influences colon carcinogenesis. Furthermore, dietary deficiencies could alter sensitivity to genetic damage and influence carcinogen metabolism contributing to CRC. High alcohol consumption increases the risk of mutations including the fact that acetaldehyde, an ethanol metabolite, is classified as a group 1 carcinogen. Tobacco smoke exposure is also a risk factor for cancer development; approximately 20% of CRCs are associated with smoking. Additionally, obese patients have a higher risk of cancer development, which is further supported by the fact that physical activity decreases CRC risk by 55%. Similarly, chronic inflammatory conditions also increase the risk of CRC development. Moreover, the circadian clock alters digestion and regulates other biochemical, physiological, and behavioral processes that could influence CRC. Taken together, colon carcinogenesis involves a number of etiological factors, and therefore, to create effective preventive strategies, molecular targets need to be identified and beleaguered prior to disease progression. With this in mind, the following is a comprehensive review identifying downstream target proteins of the above lifestyle risk factors, which are modulated during colon carcinogenesis and could be targeted for CRC prevention by novel agents including phytochemicals.
Keywords: colorectal cancer; grape seed extract; lifestyle modification; molecular targets; phytochemicals; prevention; silibinin.
Figures
References
-
- Agarwal C., Singh R. P., Dhanalakshmi S., Tyagi A. K., Tecklenburg M., Sclafani R. A., et al. (2003). Silibinin upregulates the expression of cyclin-dependent kinase inhibitors and causes cell cycle arrest and apoptosis in human colon carcinoma HT-29 cells. Oncogene 22, 8271–8282 10.1038/sj.onc.1207158 - DOI - PubMed
-
- Akagi Y., Liu W., Zebrowski B., Xie K., Ellis L. M. (1998). Regulation of vascular endothelial growth factor expression in human colon cancer by insulin-like growth factor-I. Cancer Res. 58, 4008–4014 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources