

Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology
- PMID: 23677009
- PMCID: PMC3965309
- DOI: 10.1038/ismej.2013.78
Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology
Abstract
The recent development of metaproteomics has enabled the direct identification and quantification of expressed proteins from microbial communities in situ, without the need for microbial enrichment. This became possible by (1) significant increases in quality and quantity of metagenome data and by improvements of (2) accuracy and (3) sensitivity of modern mass spectrometers (MS). The identification of physiologically relevant enzymes can help to understand the role of specific species within a community or an ecological niche. Beside identification, relative and absolute quantitation is also crucial. We will review label-free and label-based methods of quantitation in MS-based proteome analysis and the contribution of quantitative proteome data to microbial ecology. Additionally, approaches of protein-based stable isotope probing (protein-SIP) for deciphering community structures are reviewed. Information on the species-specific metabolic activity can be obtained when substrates or nutrients are labeled with stable isotopes in a protein-SIP approach. The stable isotopes ((13)C, (15)N, (36)S) are incorporated into proteins and the rate of incorporation can be used for assessing the metabolic activity of the corresponding species. We will focus on the relevance of the metabolic and phylogenetic information retrieved with protein-SIP studies and for detecting and quantifying the carbon flux within microbial consortia. Furthermore, the combination of protein-SIP with established tools in microbial ecology such as other stable isotope probing techniques are discussed.
Figures
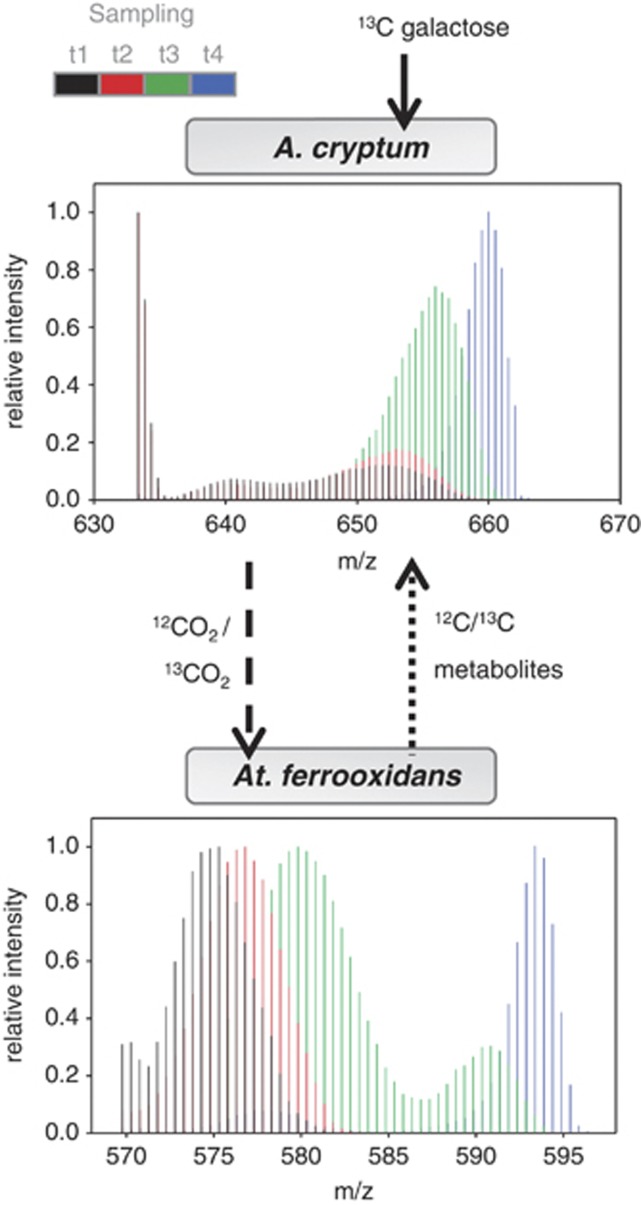
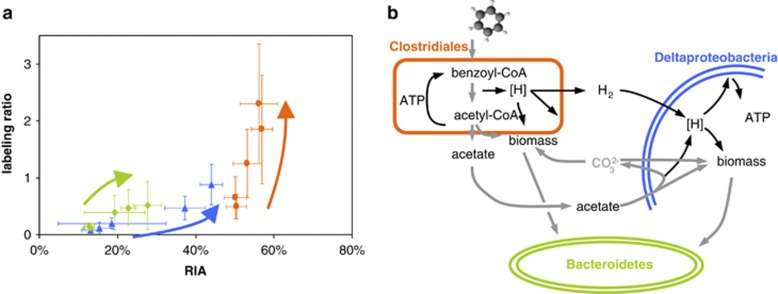
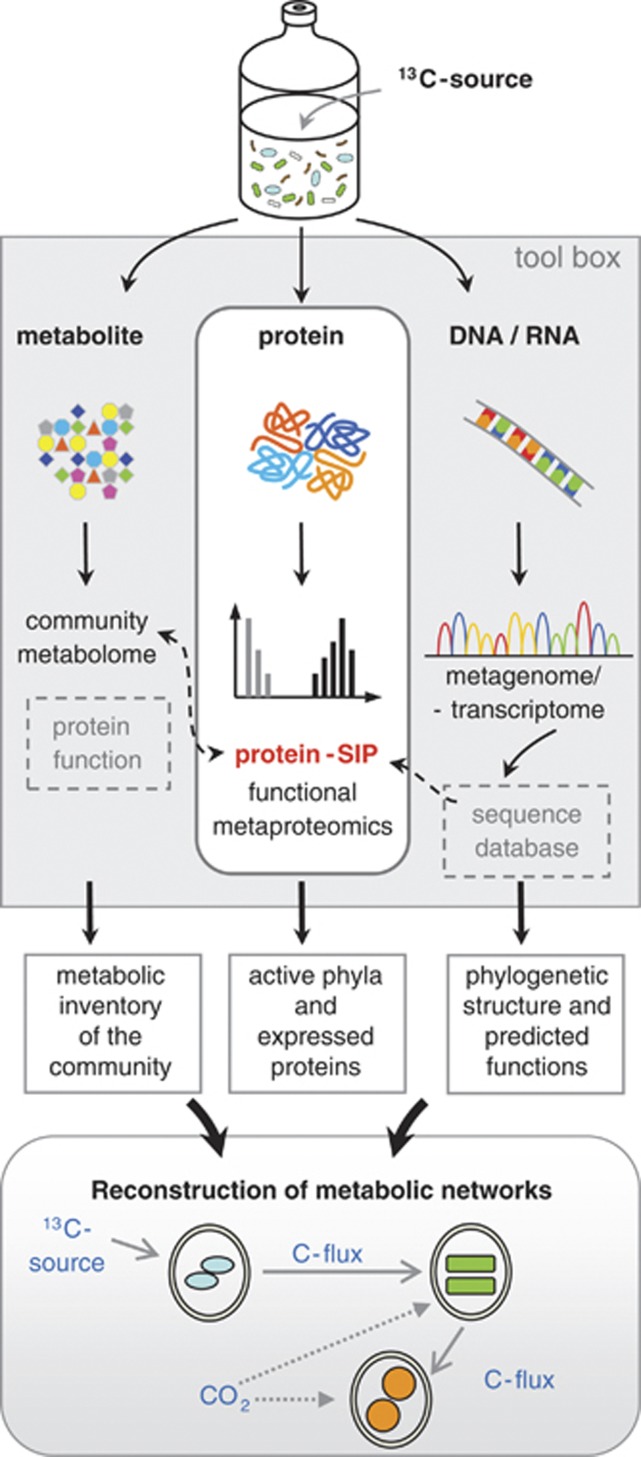
References
-
- Bastida F, Rosell M, Franchini AG, Seifert J, Finsterbusch S, Jehmlich N, et al. Elucidating MTBE degradation in a mixed consortium using a multidisciplinary approach. FEMS Microbiol Ecol. 2010;73:370–384. - PubMed
-
- Belnap CP, Pan C, VerBerkmoes NC, Power ME, Samatova NF, Carver RL, et al. Cultivation and quantitative proteomic analyses of acidophilic microbial communities. ISMEJ. 2010;4:520–530. - PubMed
-
- Beynon RJ, Pratt JM. Metabolic labeling of proteins for proteomics. Mol Cell Proteomics. 2005;4:857–872. - PubMed
-
- Bozinovski D, Herrmann S, Richnow HH, von Bergen M, Seifert J, Vogt C. Functional analysis of an anaerobic m-xylene-degrading enrichment culture using protein-based stable isotope probing. FEMS Microbiol Ecol. 2012;81:134–144. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
