

Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome
- PMID: 23677916
- PMCID: PMC3760222
- DOI: 10.1161/CIRCGENETICS.113.000138
Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome
Abstract
Background: Long QT syndrome (LQTS) is the most common cardiac channelopathy with 15 elucidated LQTS-susceptibility genes. Approximately 20% of LQTS cases remain genetically elusive.
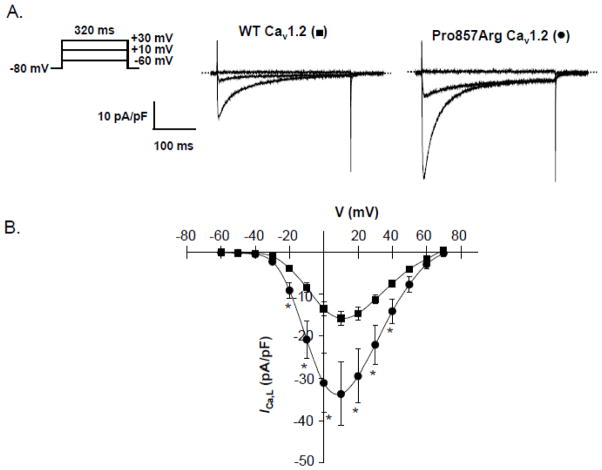
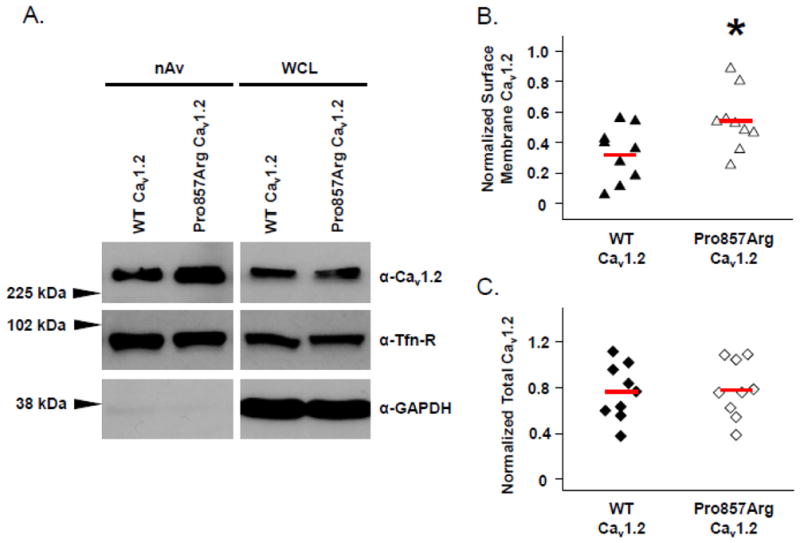
Methods and results: We combined whole-exome sequencing and bioinformatic/systems biology to identify the pathogenic substrate responsible for nonsyndromic, genotype-negative, autosomal dominant LQTS in a multigenerational pedigree, and we established the spectrum and prevalence of variants in the elucidated gene among a cohort of 102 unrelated patients with "genotype-negative/phenotype-positive" LQTS. Whole-exome sequencing was used on 3 members within a genotype-negative/phenotype-positive family. Genomic triangulation combined with bioinformatic tools and ranking algorithms led to the identification of a CACNA1C mutation. This mutation, Pro857Arg-CACNA1C, cosegregated with the disease within the pedigree, was ranked by 3 disease-network algorithms as the most probable LQTS-susceptibility gene and involves a conserved residue localizing to the proline, gltamic acid, serine, and threonine (PEST) domain in the II-III linker. Functional studies reveal that Pro857Arg-CACNA1C leads to a gain of function with increased ICa,L and increased surface membrane expression of the channel compared to wild type. Subsequent mutational analysis identified 3 additional variants within CACNA1C in our cohort of 102 unrelated cases of genotype-negative/phenotype-positive LQTS. Two of these variants also involve conserved residues within Cav1.2's PEST domain.
Conclusions: This study provides evidence that coupling whole-exome sequencing and bioinformatic/systems biology is an effective strategy for the identification of potential disease-causing genes/mutations. The identification of a functional CACNA1C mutation cosegregating with disease in a single pedigree suggests that CACNA1C perturbations may underlie autosomal dominant LQTS in the absence of Timothy syndrome.
Conflict of interest statement
Figures


Comment in
-
Taming rare variation with known biology in long QT syndrome.Circ Cardiovasc Genet. 2013 Jun;6(3):227-9. doi: 10.1161/CIRCGENETICS.113.000199. Circ Cardiovasc Genet. 2013. PMID: 23778589 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
