

Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation
- PMID: 23677989
- PMCID: PMC3689967
- DOI: 10.1074/jbc.M112.446385
Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation
Abstract
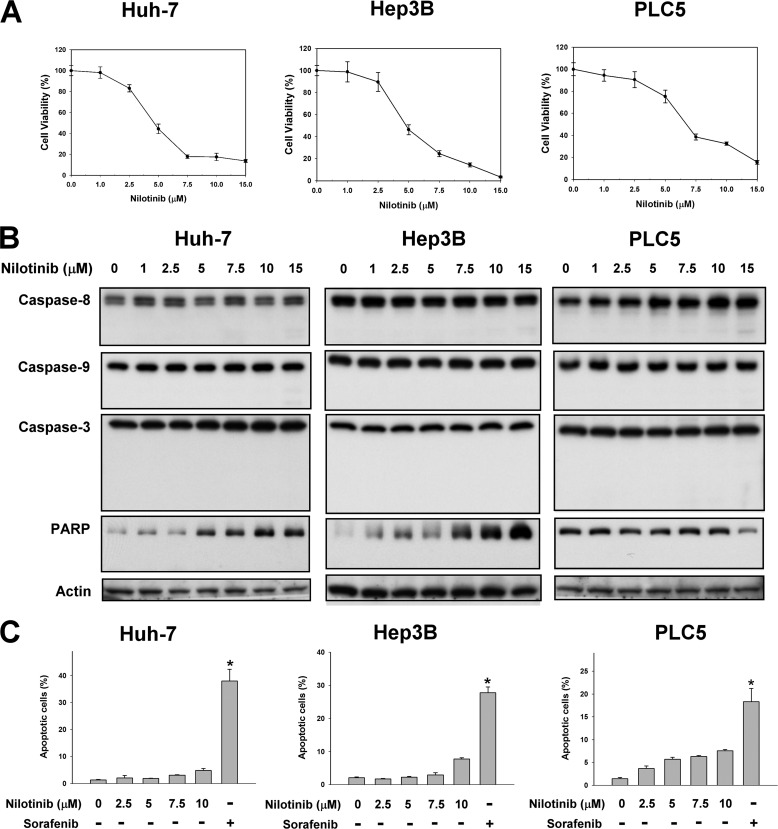
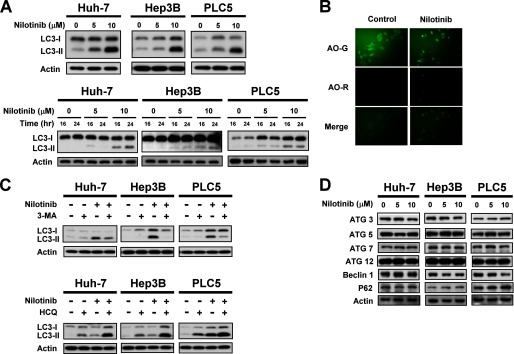
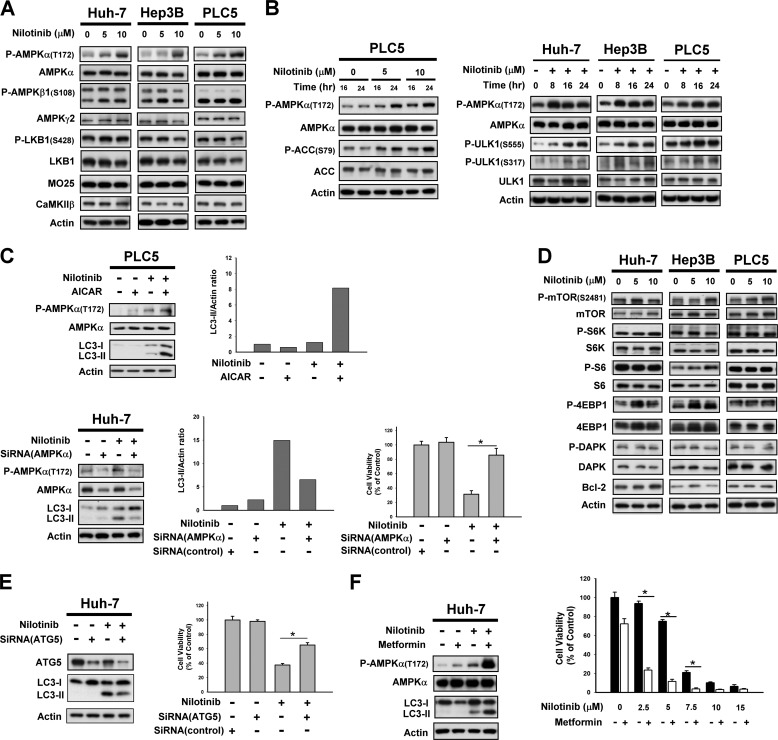
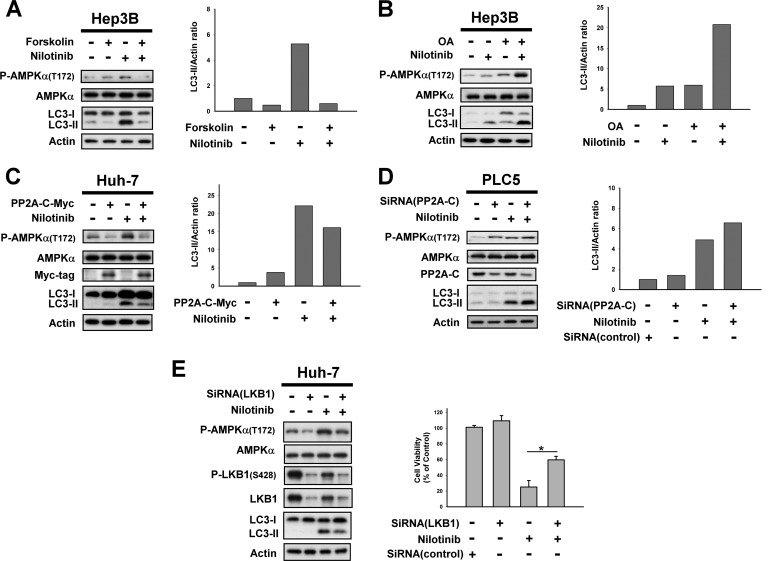
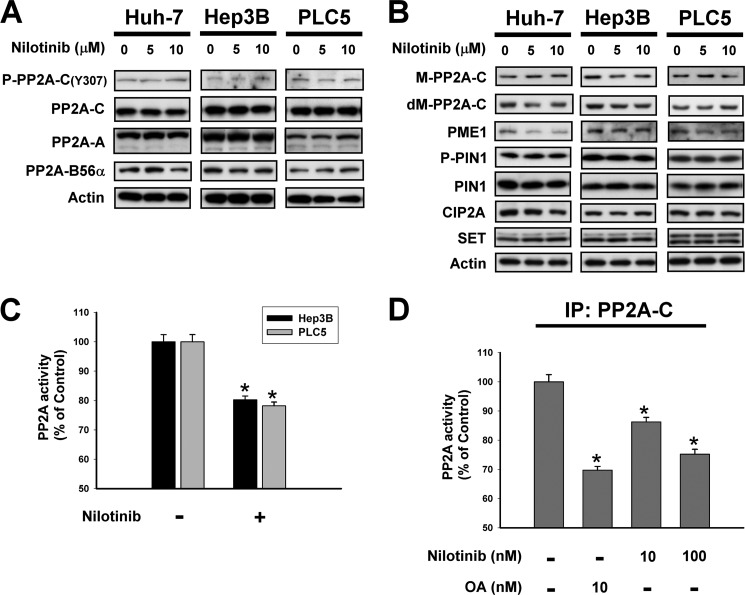
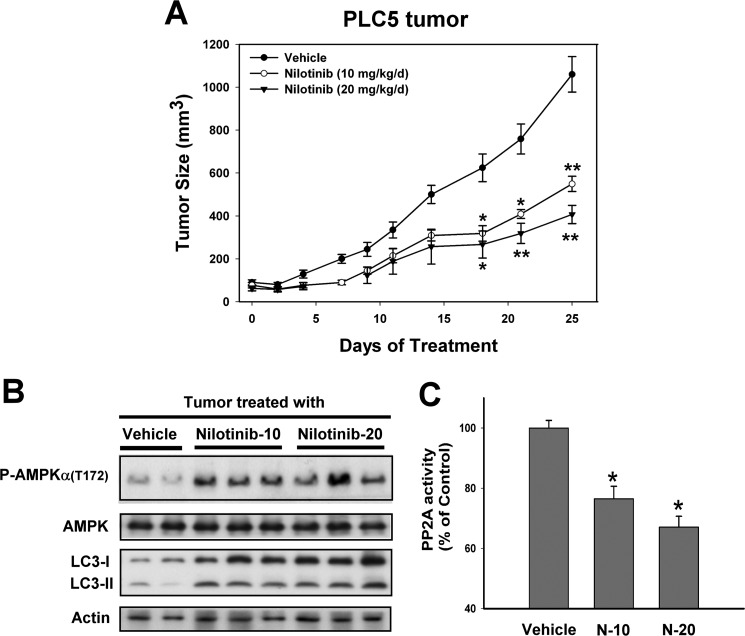
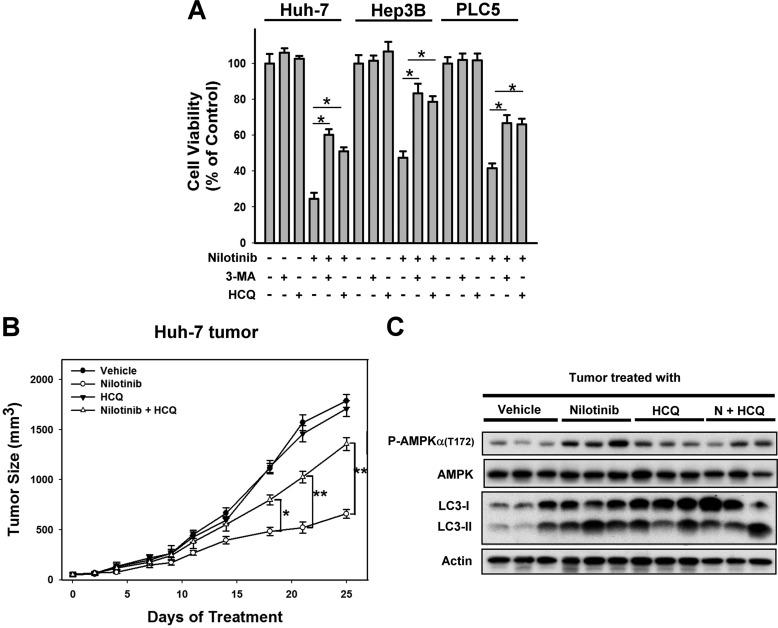
Hepatocellular carcinoma (HCC) is the most common liver cancer and the third-leading cause of cancer death worldwide. Nilotinib is an orally available receptor tyrosine kinase inhibitor approved for chronic myelogenous leukemia. This study investigated the effect of nilotinib on HCC. Nilotinib did not induce cellular apoptosis. Instead, staining with acridine orange and microtubule-associated protein 1 light chain 3 revealed that nilotinib induced autophagy in a dose- and time-dependent manner in HCC cell lines, including PLC5, Huh-7, and Hep3B. Moreover, nilotinib up-regulated the phosphryaltion of AMP-activated kinase (AMPK) and protein phosphatase PP2A inactivation were detected after nilotinib treatment. Up-regulating PP2A activity suppressed nilotinib-induced AMPK phosphorylation and autophagy, suggesting that PP2A mediates the effect of nilotinib on AMPK phosphorylation and autophagy. Our data indicate that nilotinib-induced AMPK activation is mediated by PP2A, and AMPK activation and subsequent autophagy might be a major mechanism of action of nilotinib. Growth of PLC5 tumor xenografts in BALB/c nude mice was inhibited by daily oral treatment with nilotinib. Western blot analysis showed both increased phospho-AMPK expression and decreased PP2A activity in vivo. Together, our results reveal that nilotinib induces autophagy, but not apoptosis in HCC, and that the autophagy-inducing activity is associated with PP2A-regulated AMPK phosphorylation.
Keywords: AMP-activated Kinase (AMPK); Apoptosis; Autophagy; HCC; Nilotinib; Serine Threonine Protein Phosphatase; Tyrosine Protein Kinase (Tyrosine Kinase).
Figures
References
-
- Adnane L., Trail P. A., Taylor I., Wilhelm S. M. (2006) Sorafenib (BAY 43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 407, 597–612 - PubMed
-
- Al-Kandari F. A., Owunwanne A., Syed G. M., Ar Marouf R., Elgazzar A. H., Shiekh M., Rizui A. M., Al-Ajmi J. A., Mohammed A. M. (2007) Regional cerebral blood flow in patients with sickle cell disease: study with single photon emission computed tomography. Ann. Nucl. Med. 21, 439–445 - PubMed
-
- Auclair D., Miller D., Yatsula V., Pickett W., Carter C., Chang Y., Zhang X., Wilkie D., Burd A., Shi H., Rocks S., Gedrich R., Abriola L., Vasavada H., Lynch M., Dumas J., Trail P. A., Wilhelm S. M. (2007) Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia 21, 439–445 - PubMed
-
- Wilhelm S., Carter C., Lynch M., Lowinger T., Dumas J., Smith R. A., Schwartz B., Simantov R., Kelley S. (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835–844 - PubMed
-
- Cheng A. L., Kang Y. K., Chen Z., Tsao C. J., Qin S., Kim J. S., Luo R., Feng J., Ye S., Yang T. S., Xu J., Sun Y., Liang H., Liu J., Wang J., Tak W. Y., Pan H., Burock K., Zou J., Voliotis D., Guan Z. (2009) Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomized, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
