

Dynamics of the antigen-binding grooves in CD1 proteins: reversible hydrophobic collapse in the lipid-free state
- PMID: 23677998
- PMCID: PMC3707654
- DOI: 10.1074/jbc.M113.470179
Dynamics of the antigen-binding grooves in CD1 proteins: reversible hydrophobic collapse in the lipid-free state
Abstract
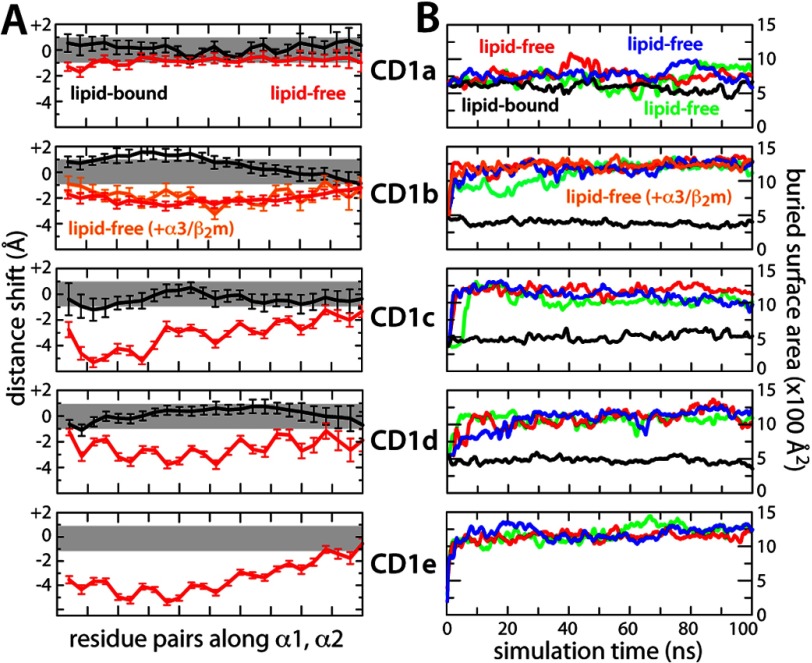
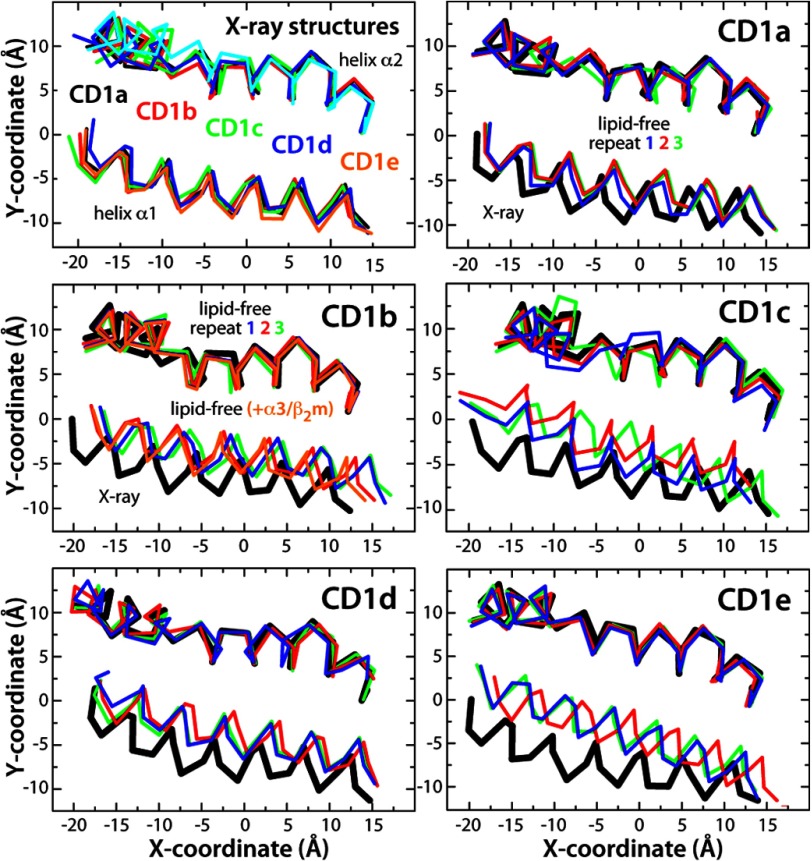
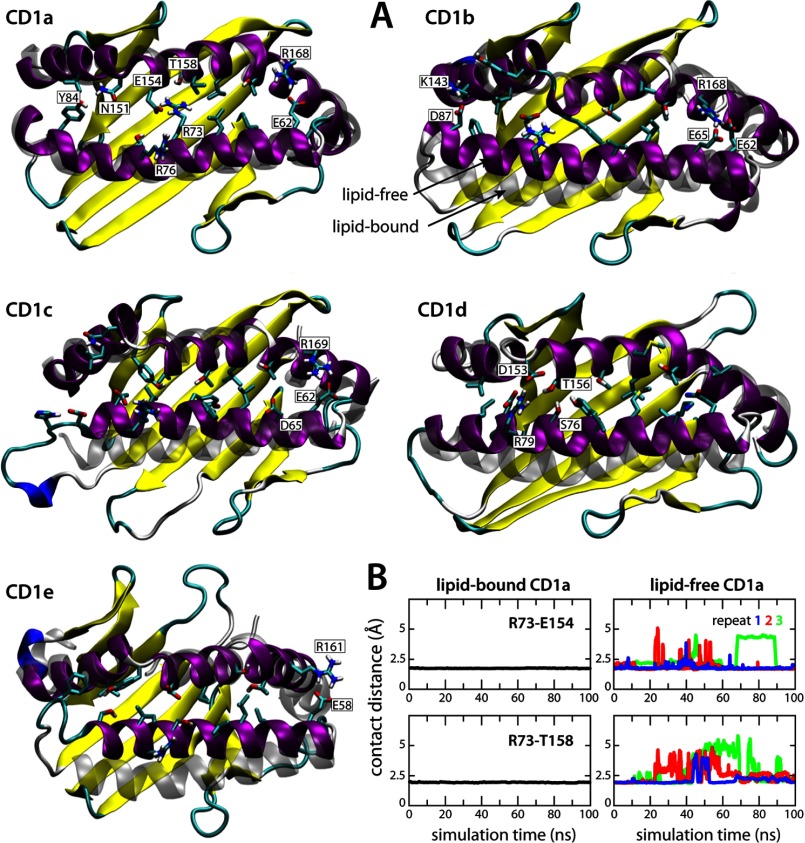
CD1 proteins mediate the presentation of endogenous and foreign lipids on the cell surface for recognition by T cell receptors. To sample a diverse antigen pool, CD1 proteins are repeatedly internalized and recycled, assisted, in some cases, by lipid transfer proteins such as saposins. The specificity of each CD1 isoform is, therefore, conferred in part by its intracellular pathway but also by distinct structural features of the antigen-binding domain. Crystal structures of CD1-lipid complexes reveal hydrophobic grooves and pockets within these binding domains that appear to be specialized for different lipids. However, the mechanism of lipid loading and release remains to be characterized. Here we gain insights into this mechanism through a meta-analysis of the five human CD1 isoforms, in the lipid-bound and lipid-free states, using all-atom molecular dynamics simulations. Strikingly, for isoforms CD1b through CD1e, our simulations show the near-complete collapse of the hydrophobic cavities in the absence of the antigen. This event results from the spontaneous closure of the binding domain entrance, flanked by two α-helices. Accordingly, we show that the anatomy of the binding cavities is restored if these α-helices are repositioned extrinsically, suggesting that helper proteins encountered during recycling facilitate lipid exchange allosterically. By contrast, we show that the binding cavity of CD1a is largely preserved in the unliganded state because of persistent electrostatic interactions that keep the portal α-helices at a constant separation. The robustness of this binding groove is consistent with the observation that lipid exchange in CD1a is not dependent on cellular internalization.
Keywords: Adaptive Immunity; Allosteric Regulation; Antigen Presentation; Cellular Immune Response; Hydrophobic Collapse; Lipid Binding Protein; Molecular Dynamics.
Figures


Similar articles
-
The molecular basis of CD1-mediated presentation of lipid antigens.Immunol Rev. 1999 Dec;172:285-96. doi: 10.1111/j.1600-065x.1999.tb01373.x. Immunol Rev. 1999. PMID: 10631954 Review.
-
CD1 assembly and the formation of CD1-antigen complexes.Curr Opin Immunol. 2005 Feb;17(1):88-94. doi: 10.1016/j.coi.2004.12.003. Curr Opin Immunol. 2005. PMID: 15653316 Review.
-
Anatomy of CD1-lipid antigen complexes.Nat Rev Immunol. 2005 May;5(5):387-99. doi: 10.1038/nri1605. Nat Rev Immunol. 2005. PMID: 15864273 Review.
-
Presentation of lipid antigens by CD1 glycoproteins.Curr Pharm Des. 2009;15(28):3311-7. doi: 10.2174/138161209789105108. Curr Pharm Des. 2009. PMID: 19860680 Free PMC article. Review.
-
The CD1 size problem: lipid antigens, ligands, and scaffolds.Cell Mol Life Sci. 2014 Aug;71(16):3069-79. doi: 10.1007/s00018-014-1603-6. Cell Mol Life Sci. 2014. PMID: 24658584 Free PMC article. Review.
Cited by
-
Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation.Chem Rev. 2019 May 8;119(9):6086-6161. doi: 10.1021/acs.chemrev.8b00608. Epub 2019 Apr 12. Chem Rev. 2019. PMID: 30978005 Free PMC article. Review.
-
Role of lipid transfer proteins in loading CD1 antigen-presenting molecules.J Lipid Res. 2018 Aug;59(8):1367-1373. doi: 10.1194/jlr.R083212. Epub 2018 Mar 19. J Lipid Res. 2018. PMID: 29559523 Free PMC article. Review.
-
Dynamic plasticity of the lipid antigen-binding site of CD1d is crucially favoured by acidic pH and helper proteins.Sci Rep. 2020 Mar 31;10(1):5714. doi: 10.1038/s41598-020-62833-y. Sci Rep. 2020. PMID: 32235847 Free PMC article.
-
Activation of human T cells by CD1 and self-lipids.Immunol Rev. 2015 Sep;267(1):16-29. doi: 10.1111/imr.12322. Immunol Rev. 2015. PMID: 26284469 Free PMC article. Review.
-
The structural basis for endotoxin-induced allosteric regulation of the Toll-like receptor 4 (TLR4) innate immune receptor.J Biol Chem. 2013 Dec 20;288(51):36215-25. doi: 10.1074/jbc.M113.501957. Epub 2013 Oct 30. J Biol Chem. 2013. PMID: 24178299 Free PMC article.
References
-
- Porcelli S. A., Segelke B. W., Sugita M., Wilson I. A., Brenner M. B. (1998) The CD1 family of lipid antigen-presenting molecules. Immunol. Today 19, 362–368 - PubMed
-
- Moody D. B., Zajonc D. M., Wilson I. A. (2005) Anatomy of CD1-lipid antigen complexes. Nat. Rev. Immunol. 5, 387–399 - PubMed
-
- Barral D. C., Brenner M. B. (2007) CD1 antigen presentation. How it works. Nat. Rev. Immunol. 7, 929–941 - PubMed
-
- Zajonc D. M., Wilson I. A. (2007) Architecture of CD1 proteins. Curr. Top. Microbiol. Immunol. 314, 27–50 - PubMed
-
- Silk J. D., Salio M., Brown J., Jones E. Y., Cerundolo V. (2008) Structural and functional aspects of lipid binding by CD1 molecules. Annu. Rev. Cell Dev. Biol. 24, 369–395 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
