

A holistic evolutionary and structural study of flaviviridae provides insights into the function and inhibition of HCV helicase
- PMID: 23678398
- PMCID: PMC3646357
- DOI: 10.7717/peerj.74
A holistic evolutionary and structural study of flaviviridae provides insights into the function and inhibition of HCV helicase
Abstract
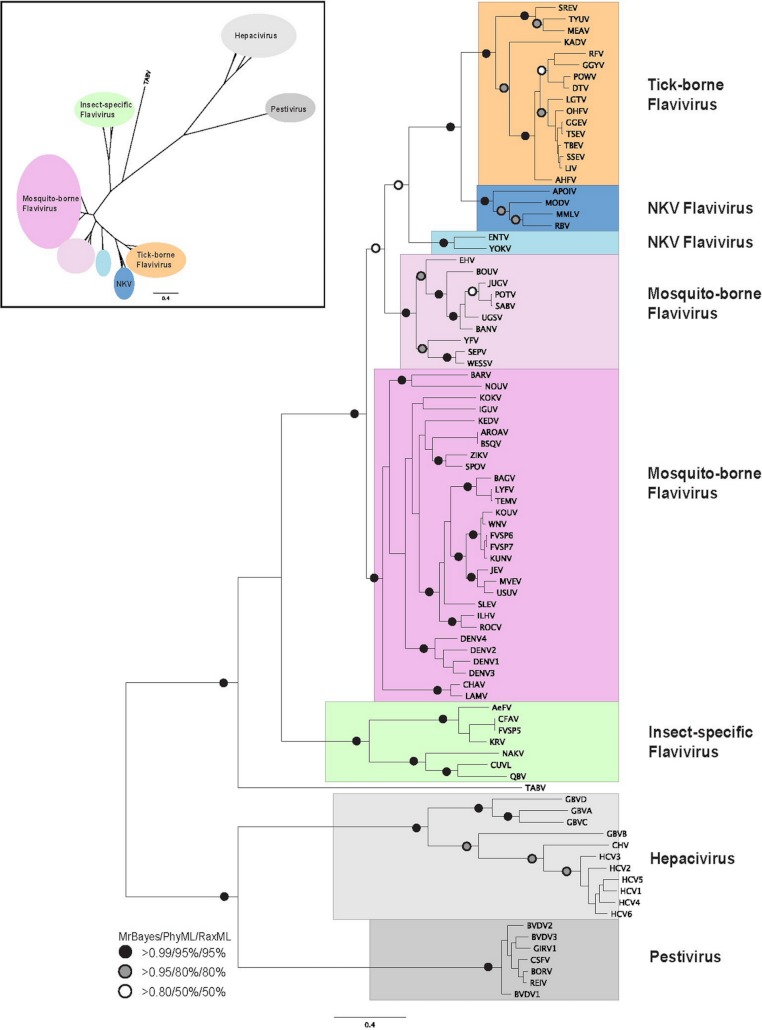
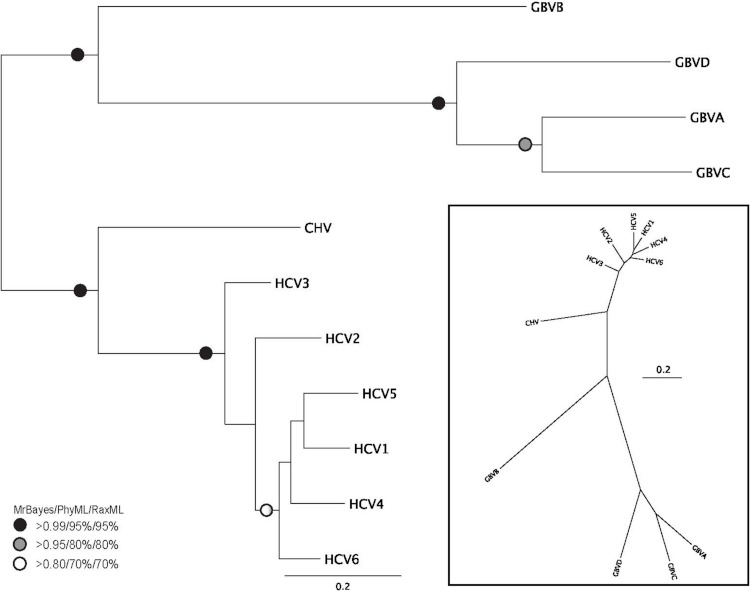
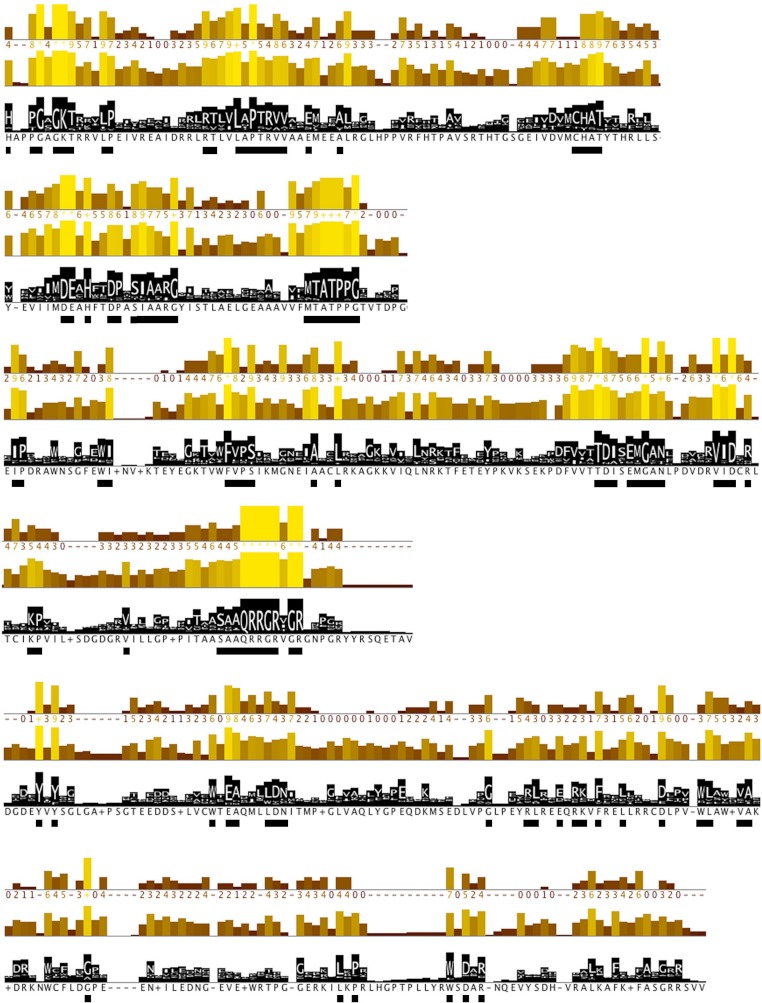
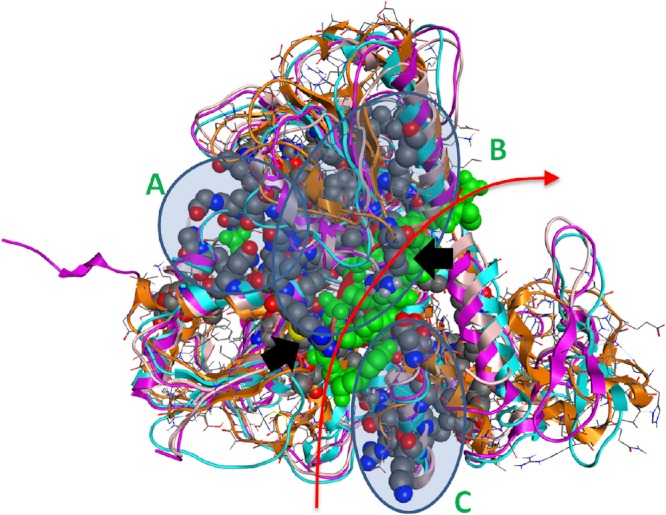
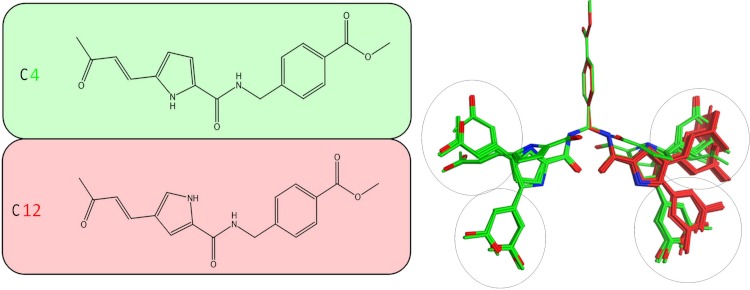
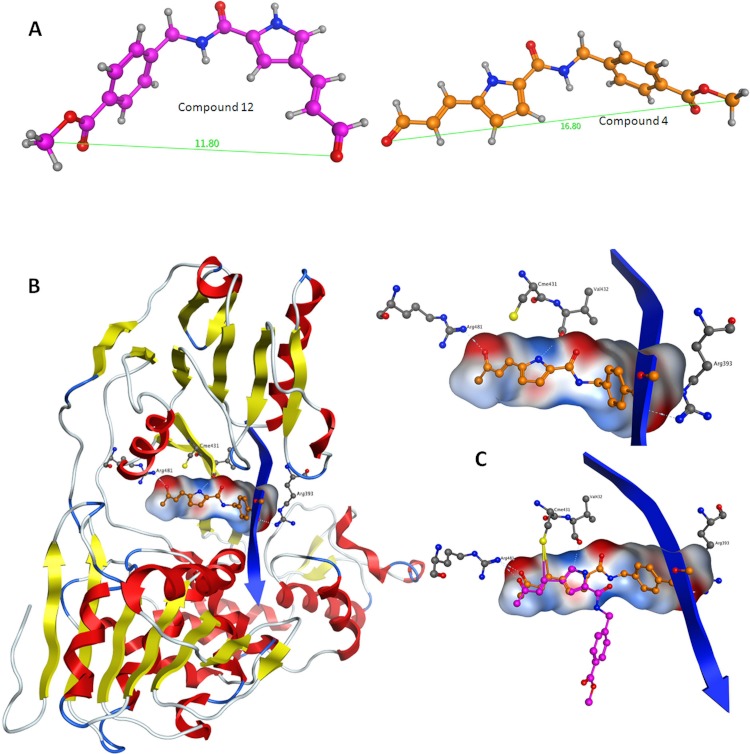
Viral RNA helicases are involved in duplex unwinding during the RNA replication of the virus. It is suggested that these helicases represent very promising antiviral targets. Viruses of the flaviviridae family are the causative agents of many common and devastating diseases, including hepatitis, yellow fever and dengue fever. As there is currently no available anti-Flaviviridae therapy, there is urgent need for the development of efficient anti-viral pharmaceutical strategies. Herein, we report the complete phylogenetic analysis across flaviviridae alongside a more in-depth evolutionary study that revealed a series of conserved and invariant amino acids that are predicted to be key to the function of the helicase. Structural molecular modelling analysis revealed the strategic significance of these residues based on their relative positioning on the 3D structures of the helicase enzymes, which may be used as pharmacological targets. We previously reported a novel series of highly potent HCV helicase inhibitors, and we now re-assess their antiviral potential using the 3D structural model of the invariant helicase residues. It was found that the most active compound of the series, compound C4, exhibited an IC50 in the submicromolar range, whereas its stereoisomer (compound C12) was completely inactive. Useful insights were obtained from molecular modelling and conformational search studies via molecular dynamics simulations. C12 tends to bend and lock in an almost "U" shape conformation, failing to establish vital interactions with the active site of HCV. On the contrary, C4 spends most of its conformational time in a straight, more rigid formation that allows it to successfully block the passage of the oligonucleotide in the ssRNA channel of the HCV helicase. This study paves the way and provides the necessary framework for the in-depth analysis required to enable the future design of new and potent anti-viral agents.
Keywords: Antiviral bioinformatics; Hepatitis C; Molecular dynamics; Structure-based drug design.
Figures
References
-
- Brancale A, Vlachaki C, Vlachakis D. Molecular modelling study of the 3D structure of the bovine viral diarrhea virus (BVDV) helicase. In Silico Biology. 2008;8(5–6):461–469. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
