

Partial cooperative unfolding in proteins as observed by hydrogen exchange mass spectrometry
- PMID: 23682200
- PMCID: PMC3652491
- DOI: 10.1080/0144235X.2012.751175
Partial cooperative unfolding in proteins as observed by hydrogen exchange mass spectrometry
Abstract
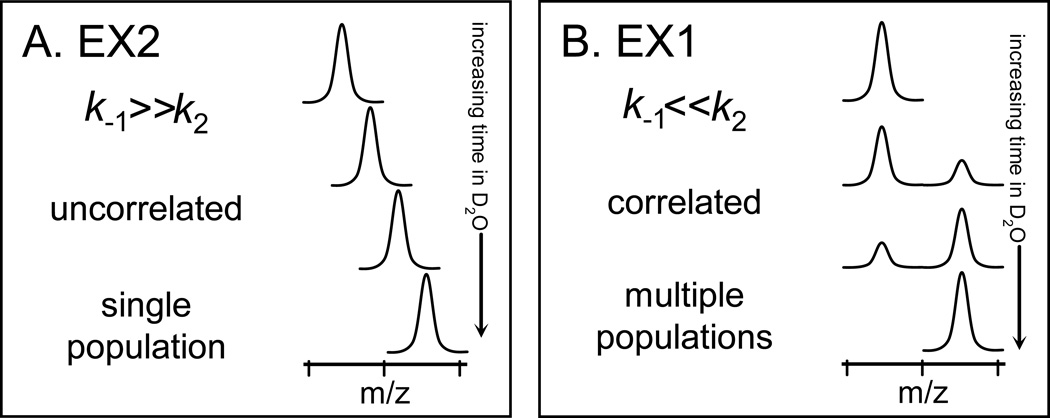
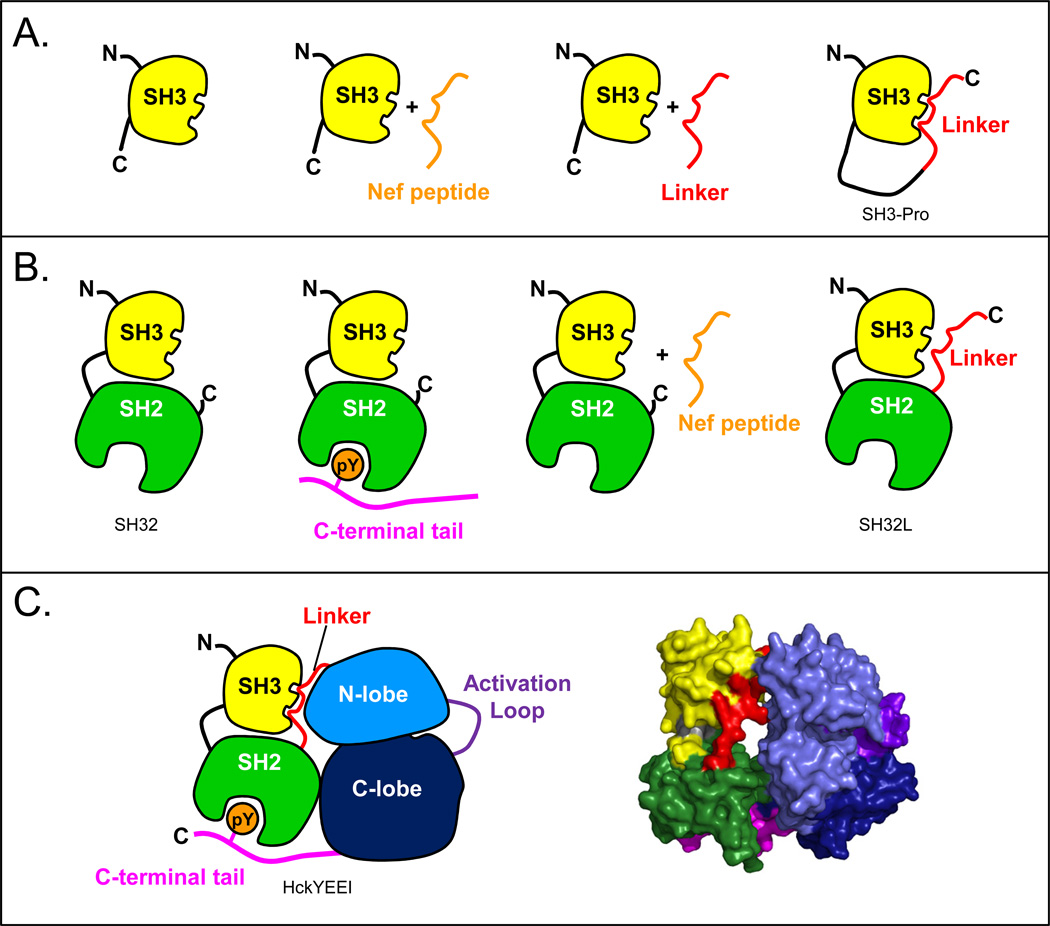
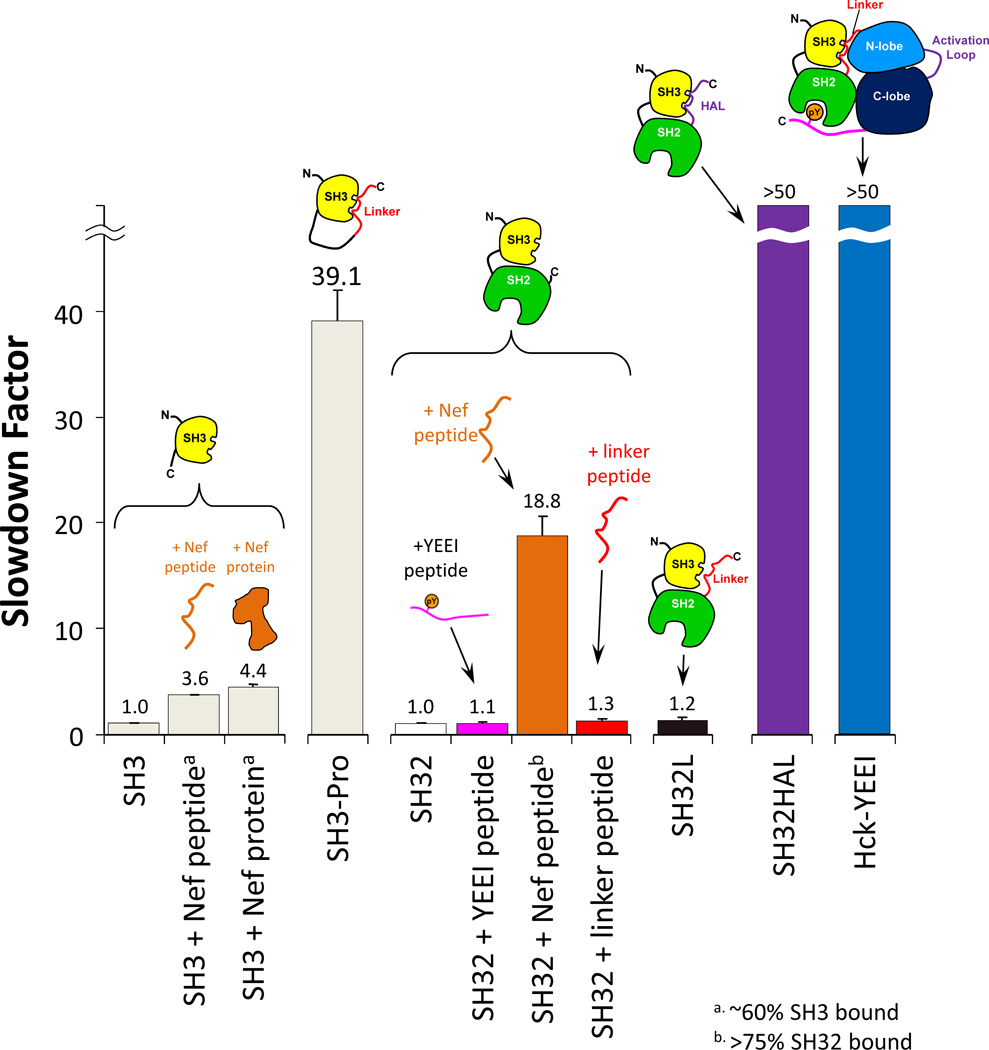
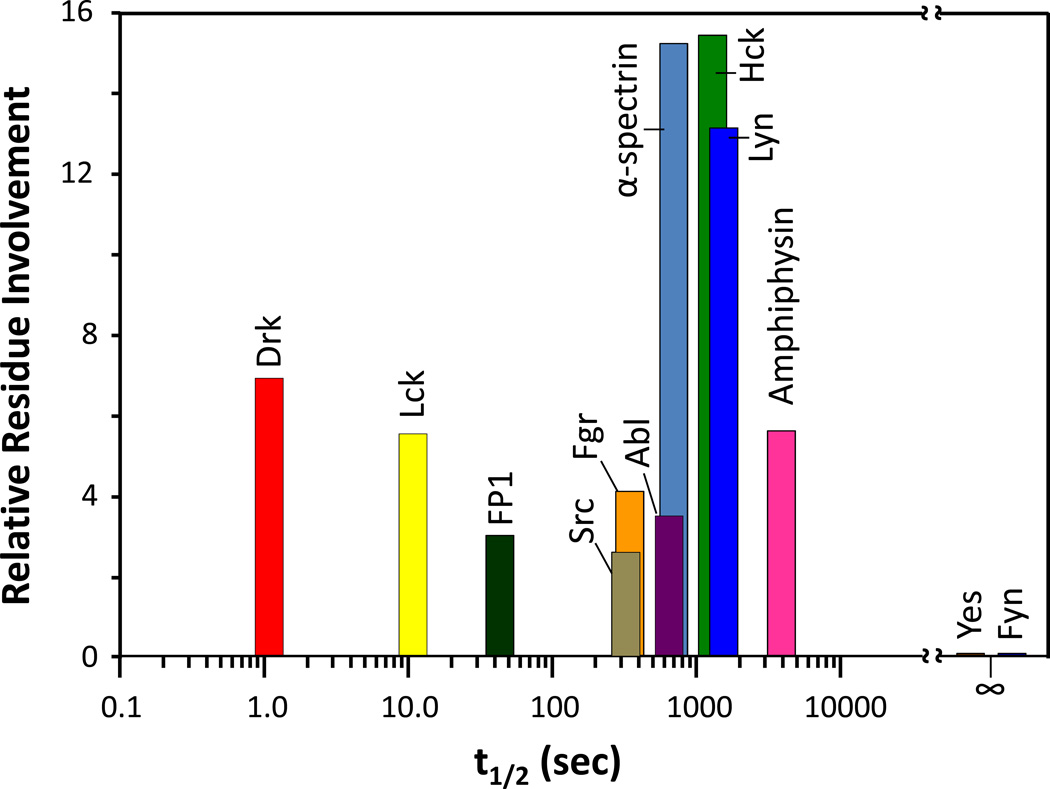
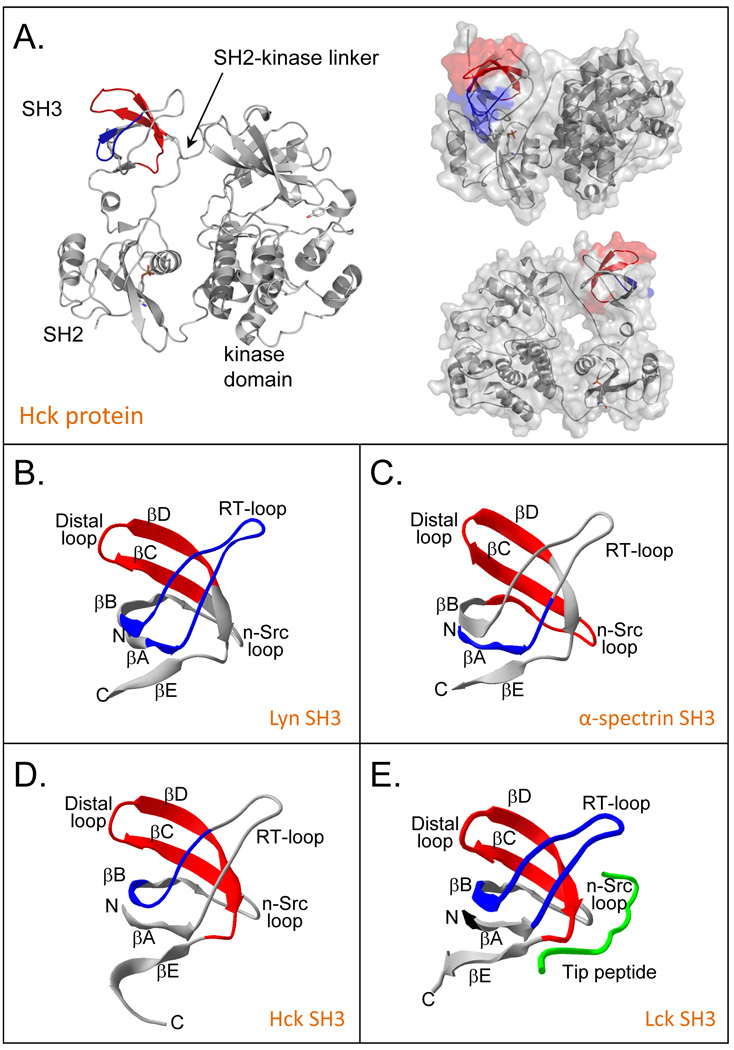
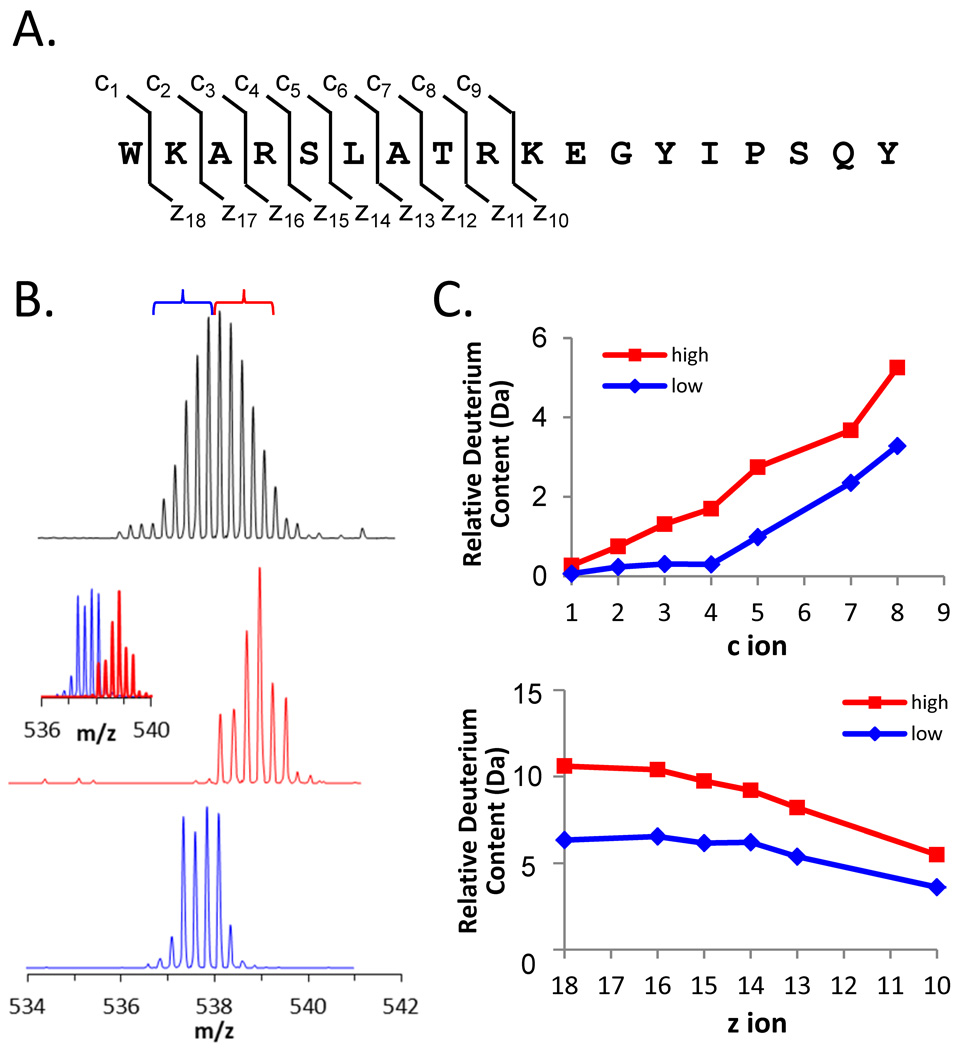
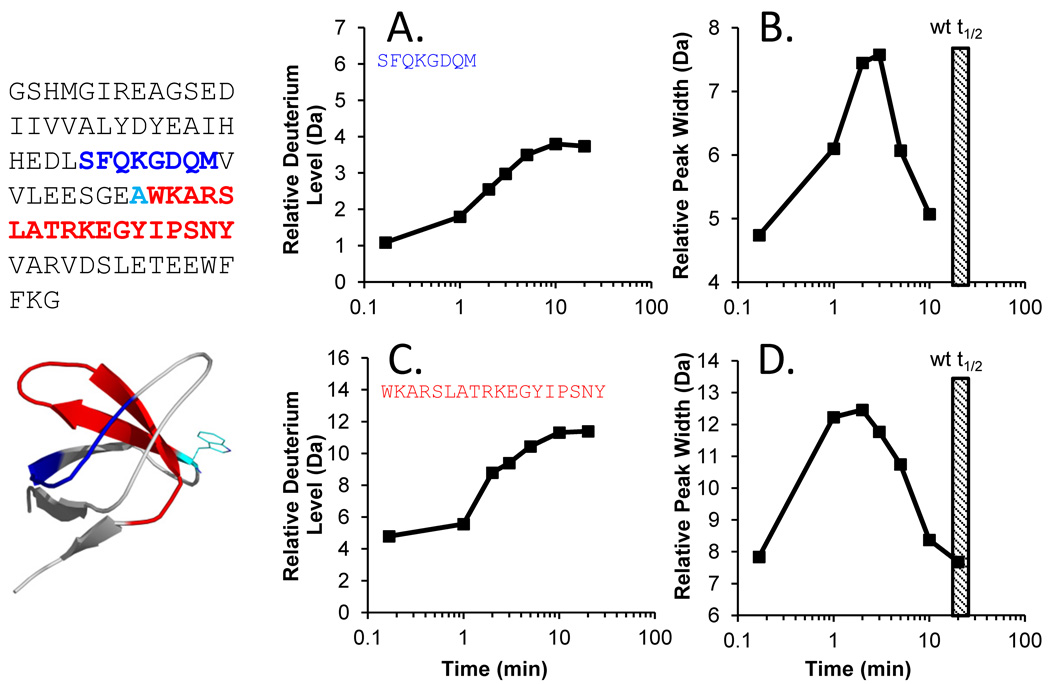
Many proteins do not exist in a single rigid conformation. Protein motions, or dynamics, exist and in many cases are important for protein function. The analysis of protein dynamics relies on biophysical techniques that can distinguish simultaneously existing populations of molecules and their rates of interconversion. Hydrogen exchange (HX) detected by mass spectrometry (MS) is contributing to our understanding of protein motions by revealing unfolding and dynamics on a wide timescale, ranging from seconds to hours to days. In this review we discuss HX MS-based analyses of protein dynamics, using our studies of multi-domain kinases as examples. Using HX MS, we have successfully probed protein dynamics and unfolding in the isolated SH3, SH2 and kinase domains of the c-Src and Abl kinase families, as well as the role of inter- and intra-molecular interactions in the global control of kinase function. Coupled with high-resolution structural information, HX MS has proved to be a powerful and versatile tool for the analysis of the conformational dynamics in these kinase systems, and has provided fresh insight regarding the regulatory control of these important signaling proteins. HX MS studies of dynamics are applicable not only to the proteins we illustrate here, but to a very wide range of proteins and protein systems, and should play a role in both classification of and greater understanding of the prevalence of protein motion.
Keywords: Abl; HDX; Hck; Lck; SH2 domain; SH3 domain; Src-family kinase; deuterium; protein dynamics, flexibility.
Figures

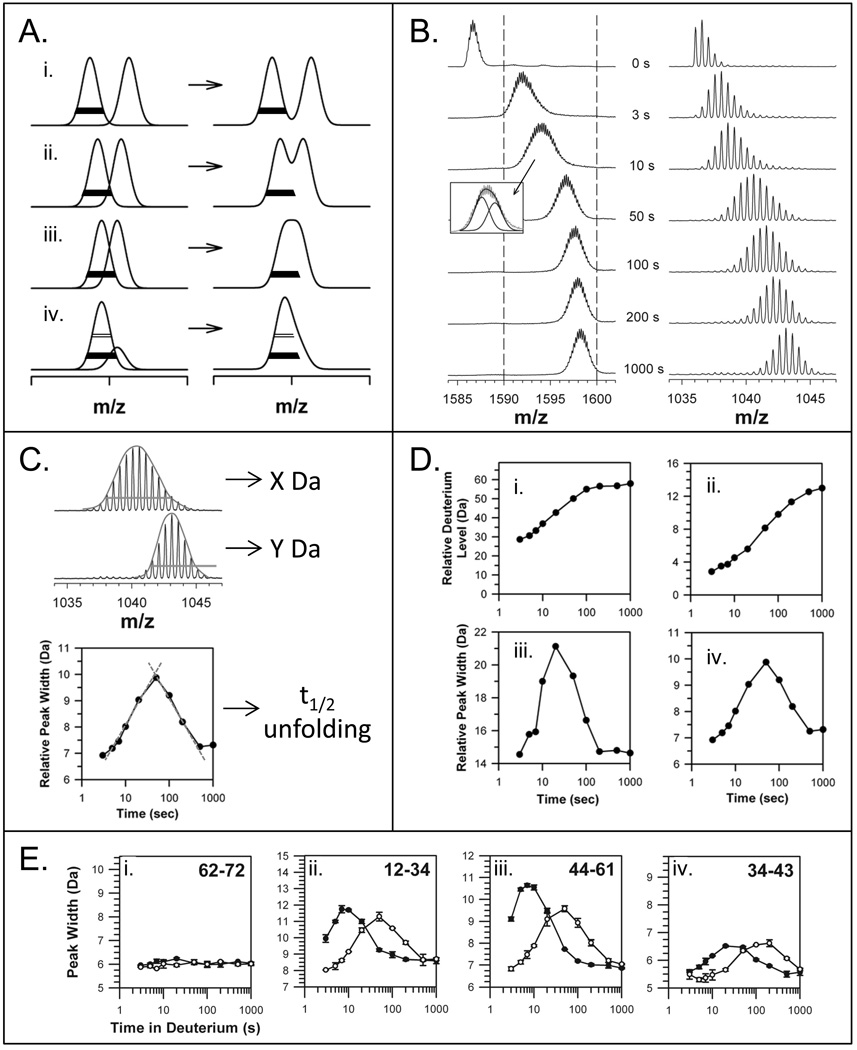
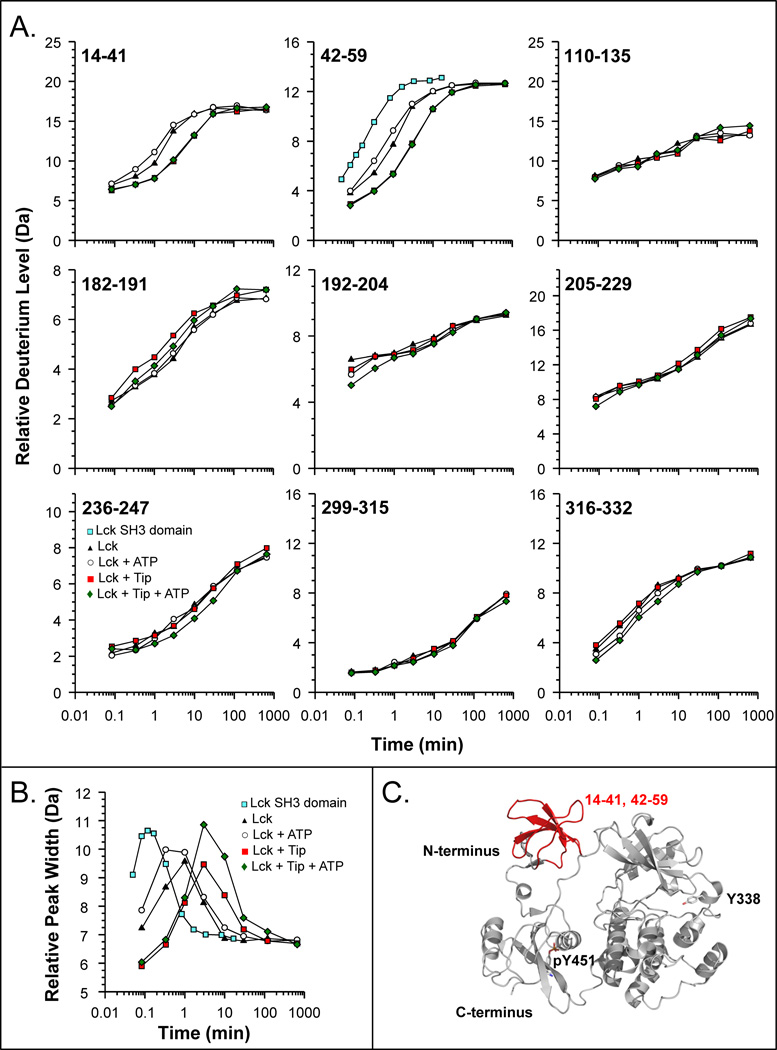
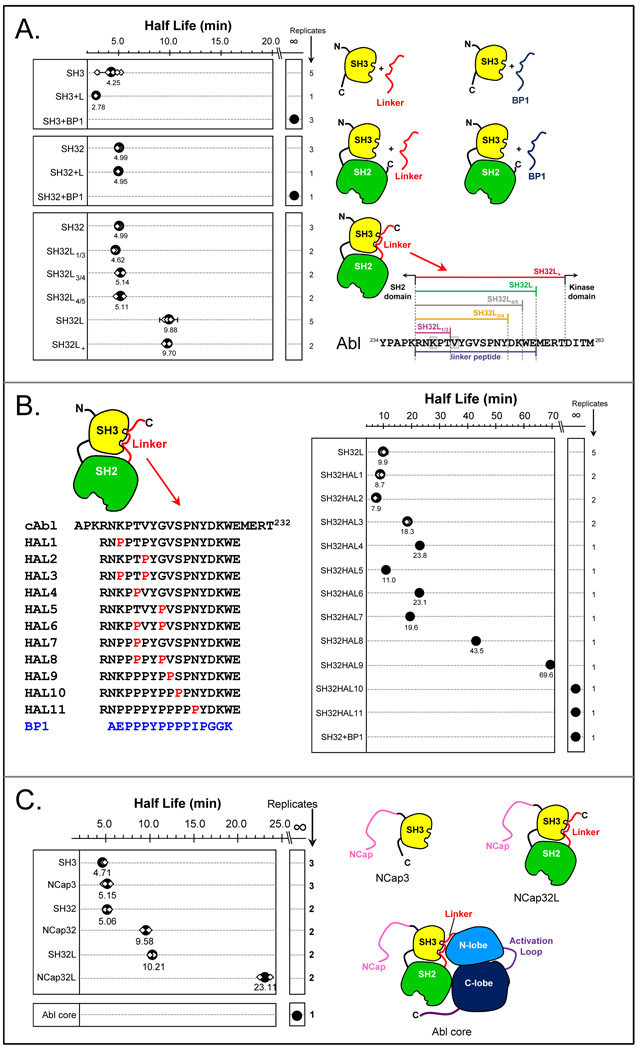
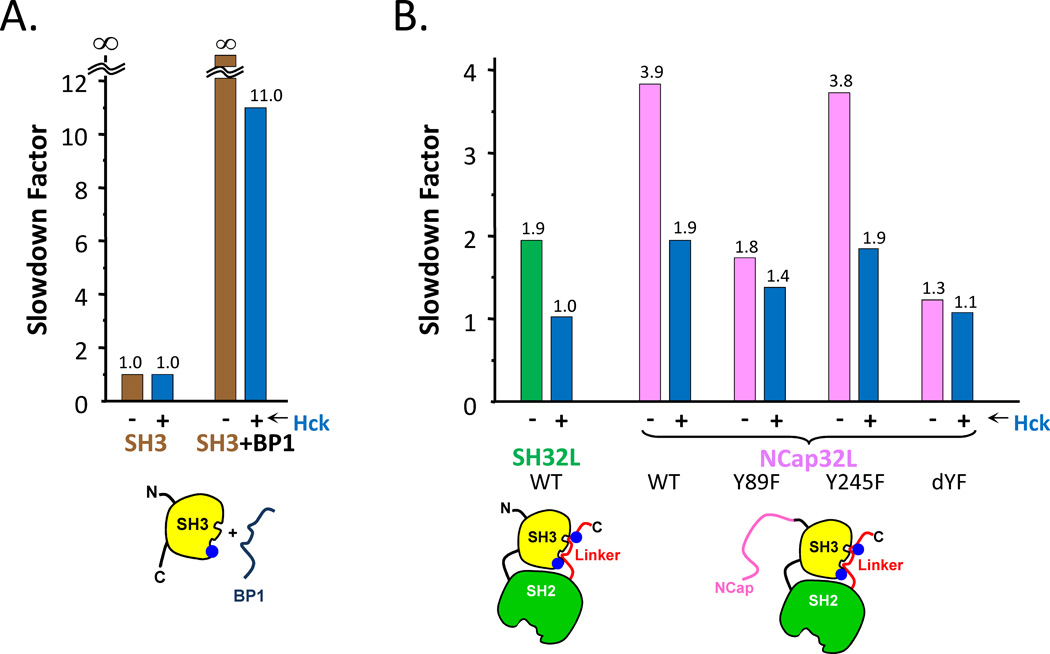
Similar articles
-
Dynamics of the Tec-family tyrosine kinase SH3 domains.Protein Sci. 2016 Apr;25(4):852-64. doi: 10.1002/pro.2887. Epub 2016 Mar 18. Protein Sci. 2016. PMID: 26808198 Free PMC article.
-
Identification and localization of slow, natural, cooperative unfolding in the hematopoietic cell kinase SH3 domain by amide hydrogen exchange and mass spectrometry.Biochemistry. 1997 Nov 25;36(47):14384-91. doi: 10.1021/bi971635m. Biochemistry. 1997. PMID: 9398156
-
Partial unfolding of diverse SH3 domains on a wide timescale.J Mol Biol. 2006 Apr 14;357(5):1592-604. doi: 10.1016/j.jmb.2006.01.075. Epub 2006 Feb 6. J Mol Biol. 2006. PMID: 16487539
-
Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: overcoming gas-phase hydrogen/deuterium scrambling.Acc Chem Res. 2014 Oct 21;47(10):3018-27. doi: 10.1021/ar500194w. Epub 2014 Aug 29. Acc Chem Res. 2014. PMID: 25171396 Review.
-
HDX-MS: An Analytical Tool to Capture Protein Motion in Action.Biomedicines. 2020 Jul 17;8(7):224. doi: 10.3390/biomedicines8070224. Biomedicines. 2020. PMID: 32709043 Free PMC article. Review.
Cited by
-
Analytical Aspects of Hydrogen Exchange Mass Spectrometry.Annu Rev Anal Chem (Palo Alto Calif). 2015;8:127-48. doi: 10.1146/annurev-anchem-062011-143113. Epub 2015 May 29. Annu Rev Anal Chem (Palo Alto Calif). 2015. PMID: 26048552 Free PMC article. Review.
-
Structure of the DDB1-AMBRA1 E3 ligase receptor complex linked to cell cycle regulation.Nat Commun. 2023 Nov 22;14(1):7631. doi: 10.1038/s41467-023-43174-6. Nat Commun. 2023. PMID: 37993427 Free PMC article.
-
QUDeX-MS: hydrogen/deuterium exchange calculation for mass spectra with resolved isotopic fine structure.BMC Bioinformatics. 2014 Dec 11;15(1):403. doi: 10.1186/s12859-014-0403-1. BMC Bioinformatics. 2014. PMID: 25495703 Free PMC article.
-
Interlaboratory Comparison of Hydrogen-Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb.Anal Chem. 2019 Jun 4;91(11):7336-7345. doi: 10.1021/acs.analchem.9b01100. Epub 2019 May 14. Anal Chem. 2019. PMID: 31045344 Free PMC article.
-
Fluorescence Polarization Screening Assays for Small Molecule Allosteric Modulators of ABL Kinase Function.PLoS One. 2015 Jul 29;10(7):e0133590. doi: 10.1371/journal.pone.0133590. eCollection 2015. PLoS One. 2015. PMID: 26222440 Free PMC article.
References
-
- Engen JR, Smithgall TE, Gmeiner WH, Smith DL. Biochemistry. 1997;36(47):14384. - PubMed
-
- Linderstrom-Lang KU, Schellman JA. In: The Enzymes. Boyer PD, Lardy H, Myrbäck K, editors. Vol. 1. New York: Academic; 1959.
-
- Berger A, Linderstrom-Lang K. Arch Biochem Biophys. 1957;69:106. - PubMed
-
- Frauenfelder H, Parak F, Young RD. Annu Rev Biophys Biophys Chem. 1988;17:451. - PubMed
-
- Frauenfelder H, Sligar SG, Wolynes PG. Science. 1991;254(5038):1598. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous