

Properties and rates of germline mutations in humans
- PMID: 23684843
- PMCID: PMC3785239
- DOI: 10.1016/j.tig.2013.04.005
Properties and rates of germline mutations in humans
Abstract
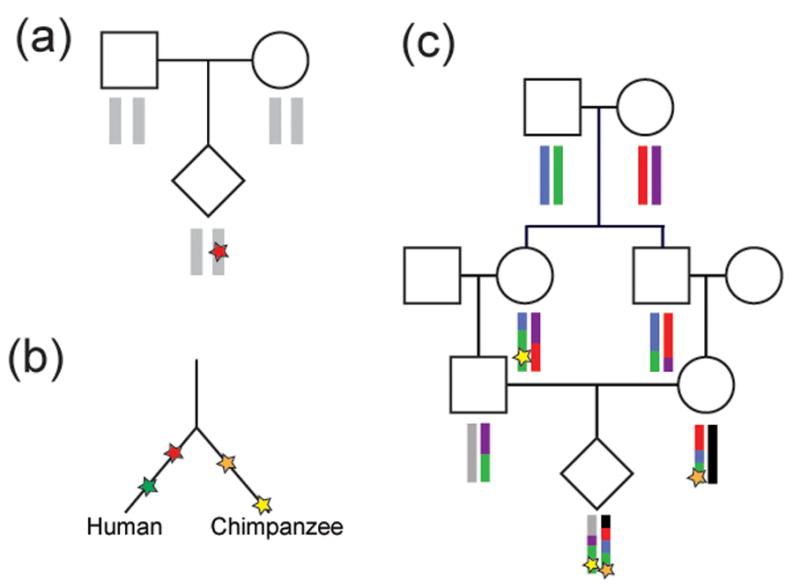
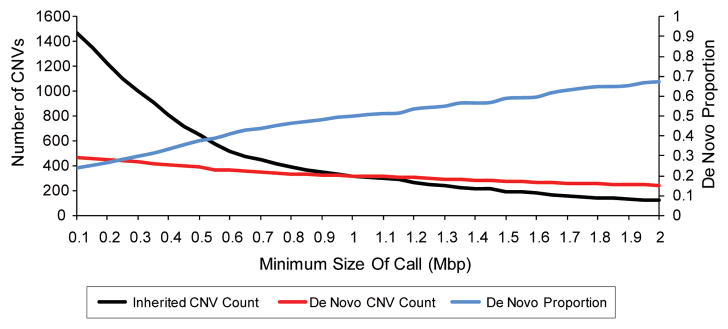
All genetic variation arises via new mutations; therefore, determining the rate and biases for different classes of mutation is essential for understanding the genetics of human disease and evolution. Decades of mutation rate analyses have focused on a relatively small number of loci because of technical limitations. However, advances in sequencing technology have allowed for empirical assessments of genome-wide rates of mutation. Recent studies have shown that 76% of new mutations originate in the paternal lineage and provide unequivocal evidence for an increase in mutation with paternal age. Although most analyses have focused on single nucleotide variants (SNVs), studies have begun to provide insight into the mutation rate for other classes of variation, including copy number variants (CNVs), microsatellites, and mobile element insertions (MEIs). Here, we review the genome-wide analyses for the mutation rate of several types of variants and suggest areas for future research.
Keywords: de novo mutation; genome wide; germline mutation rate; paternal age; paternal bias.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures



References
-
- Haldane JBS. The rate of spontaneous muation of a human gene. Journal of Genetics. 1935;31:317–326. - PubMed
-
- Haldane JB. The mutation rate of the gene for haemophilia, and its segregation ratios in males and females. Ann Eugen. 1947;13:262–271. - PubMed
-
- Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12–27. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
