

The PhyloFacts FAT-CAT web server: ortholog identification and function prediction using fast approximate tree classification
- PMID: 23685612
- PMCID: PMC3692063
- DOI: 10.1093/nar/gkt399
The PhyloFacts FAT-CAT web server: ortholog identification and function prediction using fast approximate tree classification
Abstract
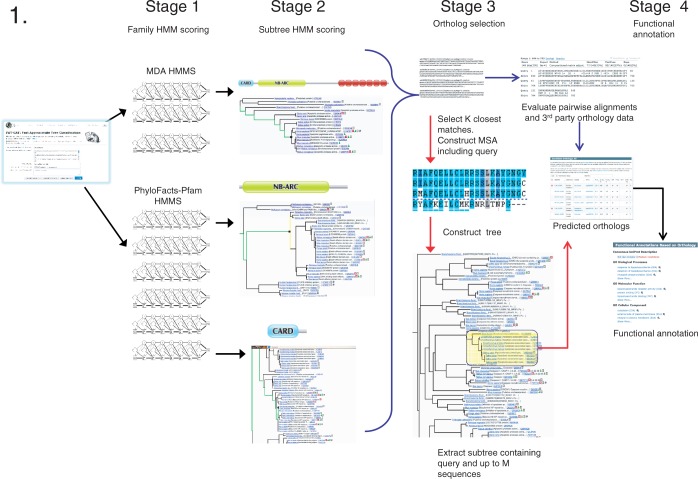
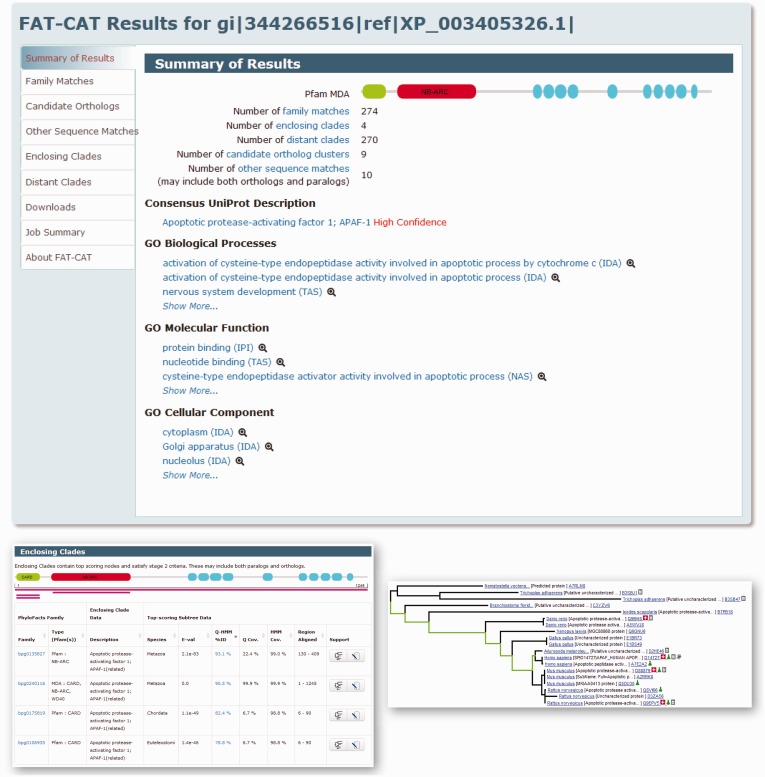
The PhyloFacts 'Fast Approximate Tree Classification' (FAT-CAT) web server provides a novel approach to ortholog identification using subtree hidden Markov model-based placement of protein sequences to phylogenomic orthology groups in the PhyloFacts database. Results on a data set of microbial, plant and animal proteins demonstrate FAT-CAT's high precision at separating orthologs and paralogs and robustness to promiscuous domains. We also present results documenting the precision of ortholog identification based on subtree hidden Markov model scoring. The FAT-CAT phylogenetic placement is used to derive a functional annotation for the query, including confidence scores and drill-down capabilities. PhyloFacts' broad taxonomic and functional coverage, with >7.3 M proteins from across the Tree of Life, enables FAT-CAT to predict orthologs and assign function for most sequence inputs. Four pipeline parameter presets are provided to handle different sequence types, including partial sequences and proteins containing promiscuous domains; users can also modify individual parameters. PhyloFacts trees matching the query can be viewed interactively online using the PhyloScope Javascript tree viewer and are hyperlinked to various external databases. The FAT-CAT web server is available at http://phylogenomics.berkeley.edu/phylofacts/fatcat/.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
