

Interplay between DNA tumor viruses and the host DNA damage response
- PMID: 23686238
- PMCID: PMC6707713
- DOI: 10.1007/978-3-642-37765-5_9
Interplay between DNA tumor viruses and the host DNA damage response
Abstract
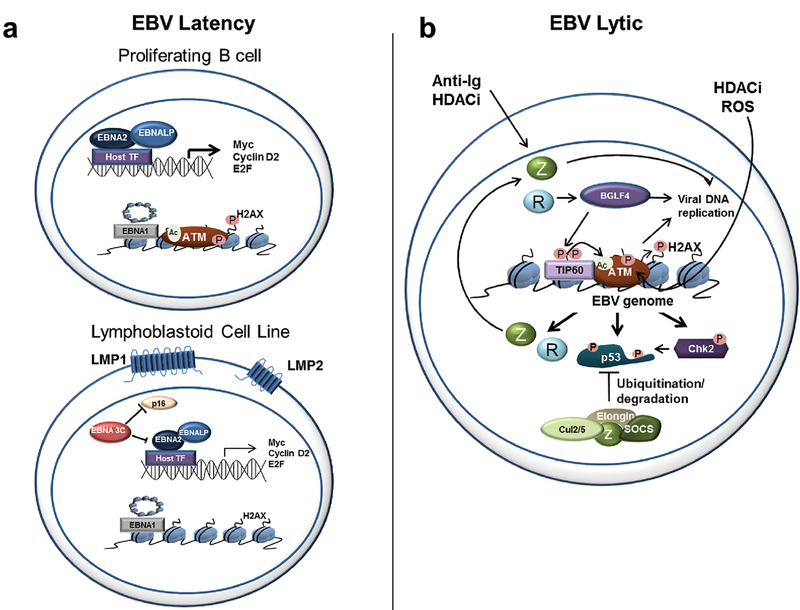
Viruses encounter many challenges within host cells in order to replicate their nucleic acid. In the case of DNA viruses, one challenge that must be overcome is recognition of viral DNA structures by the host DNA damage response (DDR) machinery. This is accomplished in elegant and unique ways by different viruses as each has specific needs and sensitivities dependent on its life cycle. In this review, we focus on three DNA tumor viruses and their interactions with the DDR. The viruses Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), and human papillomavirus (HPV) account for nearly all of the virus-associated human cancers worldwide. These viruses have also been excellent models for the study of oncogenic virus-mediated cell transformation. In this review, we will discuss how each of these viruses engage and subvert aspects of the host DDR. The first level of DDR engagement is a result of the genetic linkage between the oncogenic potential of these viruses and their ability to replicate. Namely, the promotion of cells from quiescence into the cell cycle to facilitate virus replication can be sensed through aberrant cellular DNA replication structures which activate the DDR and hinder cell transformation. DNA tumor viruses subvert this growth-suppressive DDR through changes in viral oncoprotein expression which ultimately facilitate virus replication. An additional level of DDR engagement is through direct detection of replicating viral DNA. These interactions parallel those observed in other DNA virus systems in that the need to subvert these intrinsic sensors of aberrant DNA structure in order to replicate must be in place. DNA tumor viruses are no exception. This review will cover the molecular features of DNA tumor virus interactions with the host DDR and the consequences for virus replication.
Figures



References
-
- Bartek J, Bartkova J, and Lukas J, DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene, 2007. 26(56): p. 7773–9. - PubMed
-
- Smith J, et al. , The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res, 2010. 108: p. 73–112. - PubMed
-
- Falck J, Coates J, and Jackson SP, Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature, 2005. 434(7033): p. 605–11. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
