

Deficiency of TDAG51 protects against atherosclerosis by modulating apoptosis, cholesterol efflux, and peroxiredoxin-1 expression
- PMID: 23686369
- PMCID: PMC3698773
- DOI: 10.1161/JAHA.113.000134
Deficiency of TDAG51 protects against atherosclerosis by modulating apoptosis, cholesterol efflux, and peroxiredoxin-1 expression
Abstract
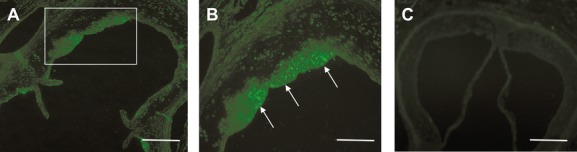
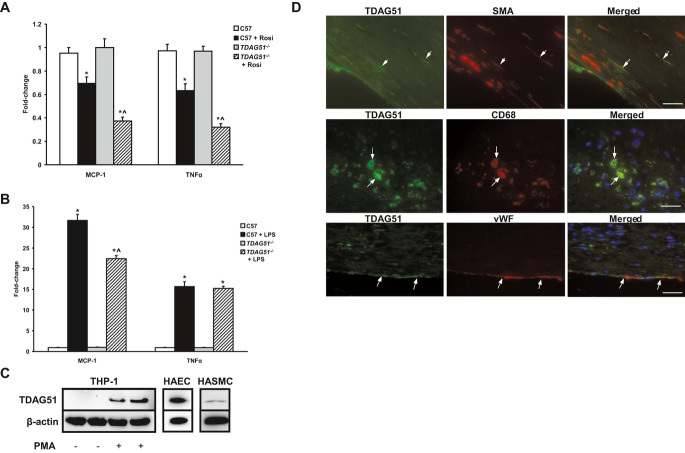
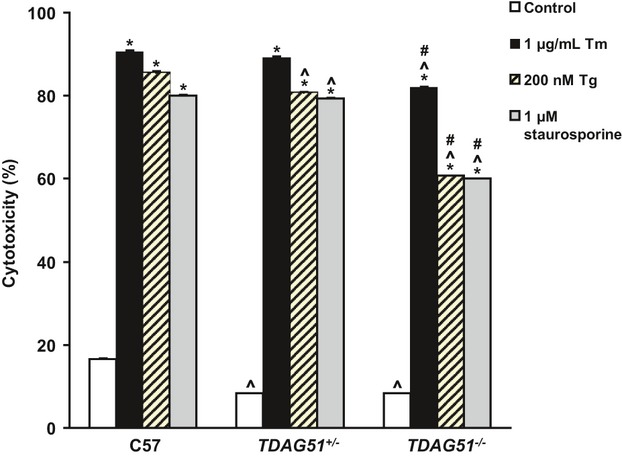
Background: Apoptosis caused by endoplasmic reticulum (ER) stress contributes to atherothrombosis, the underlying cause of cardiovascular disease (CVD). T-cell death-associated gene 51 (TDAG51), a member of the pleckstrin homology-like domain gene family, is induced by ER stress, causes apoptosis when overexpressed, and is present in lesion-resident macrophages and endothelial cells.
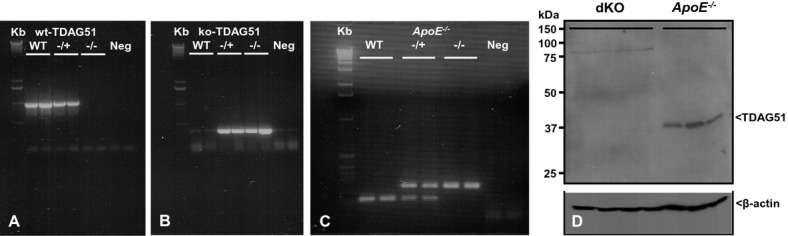
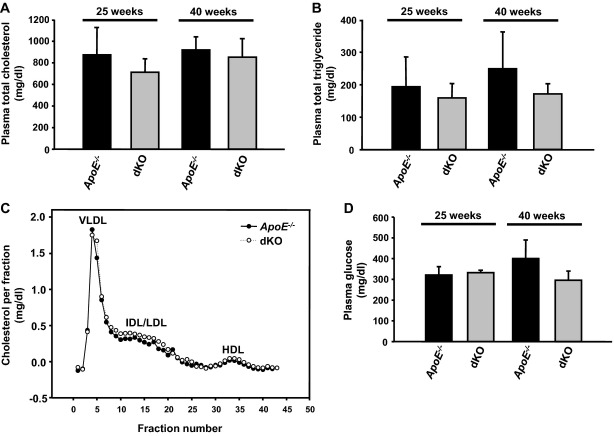
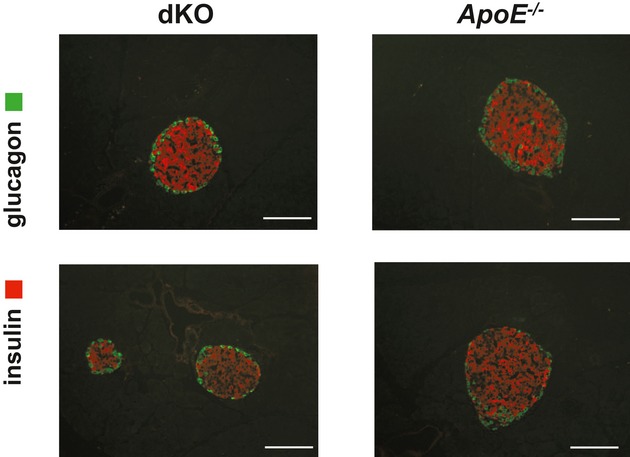
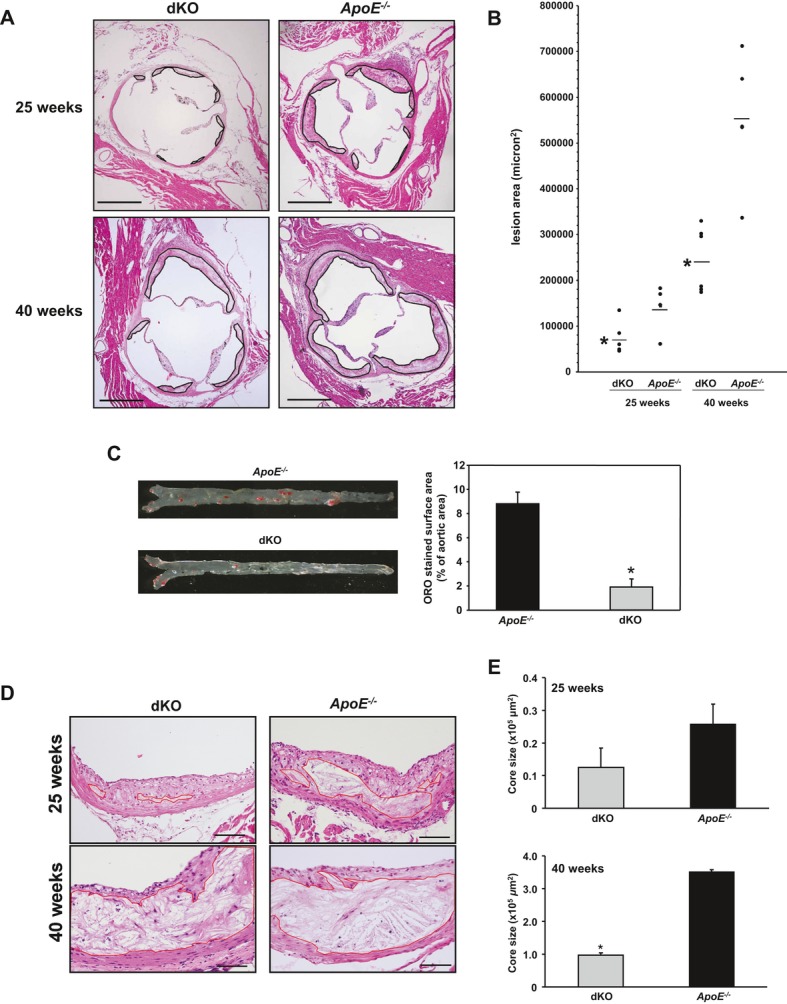
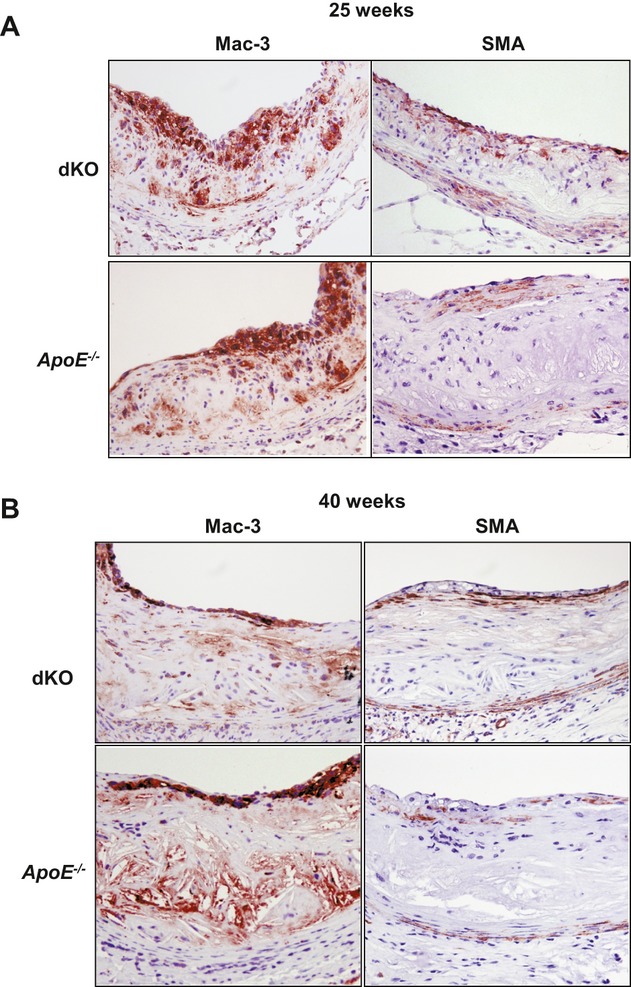
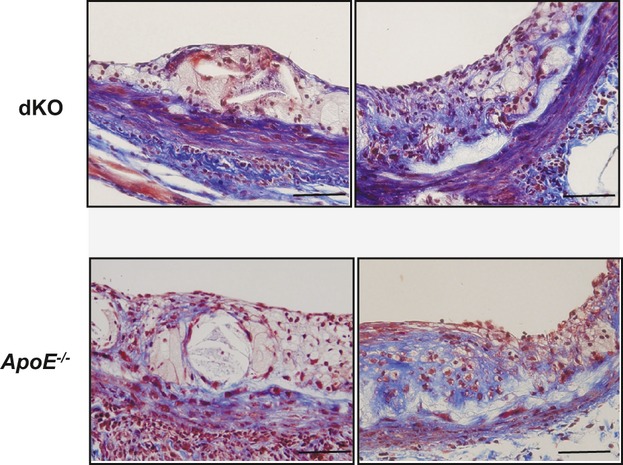
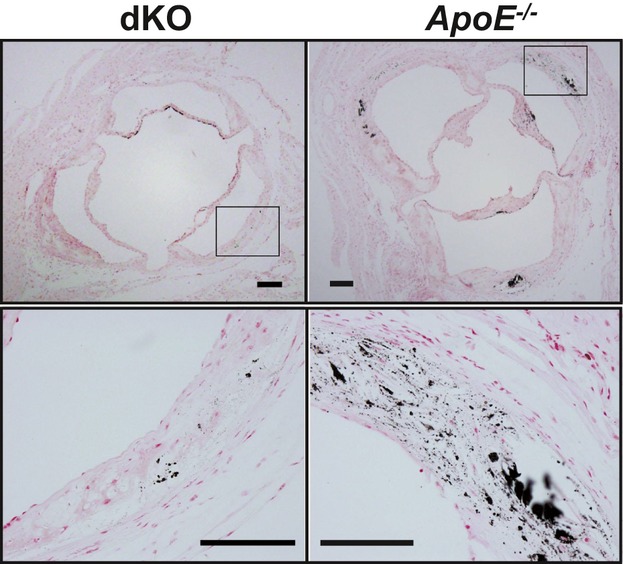
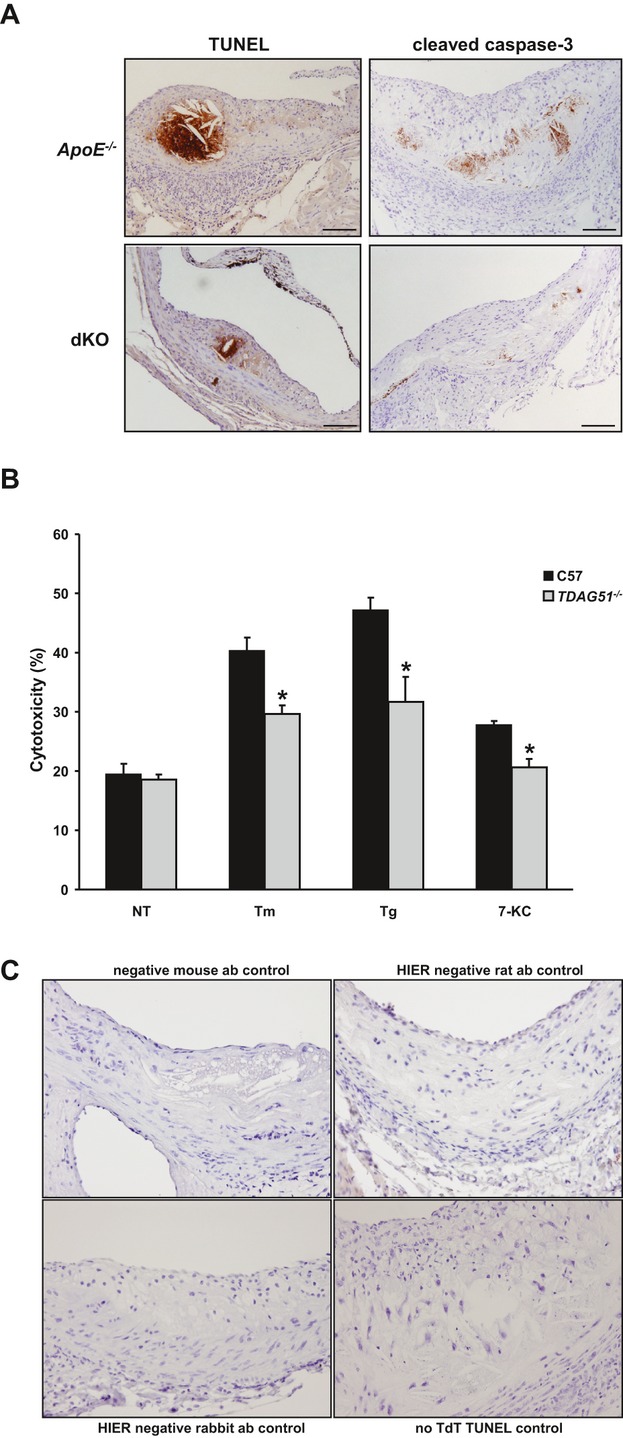
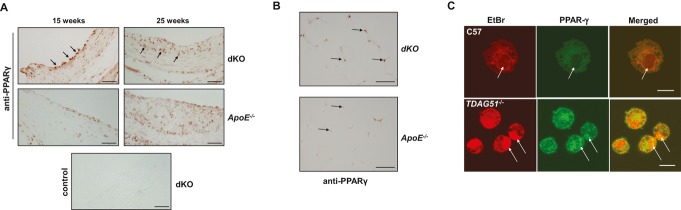
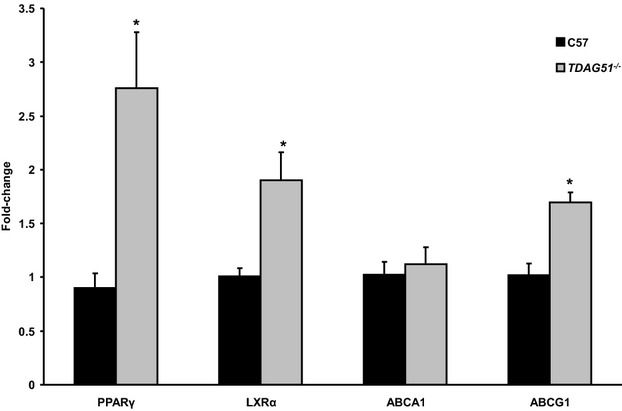
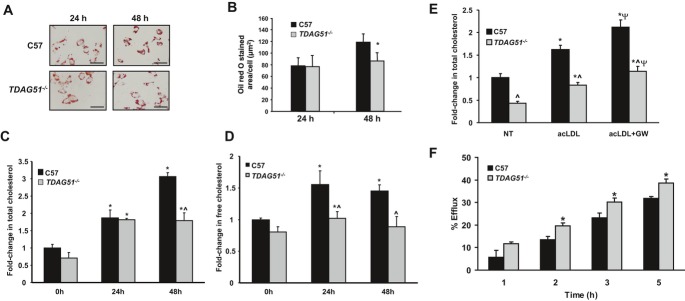
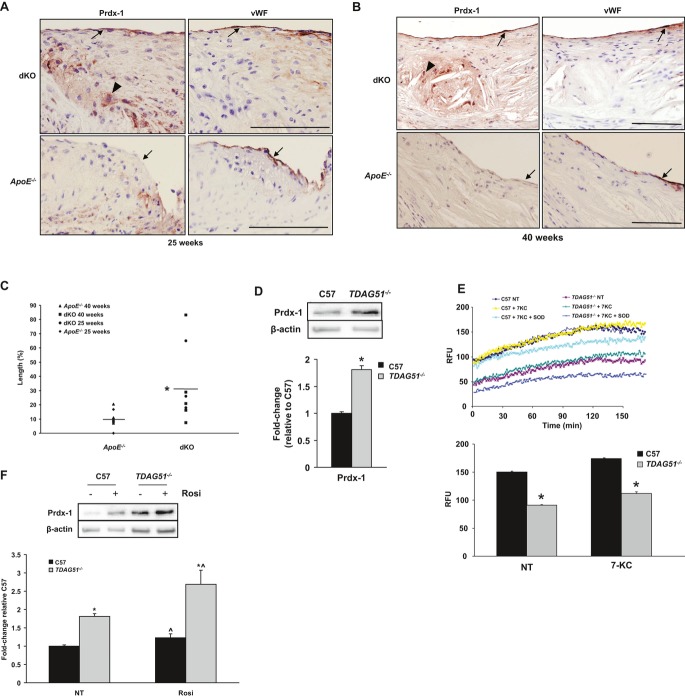
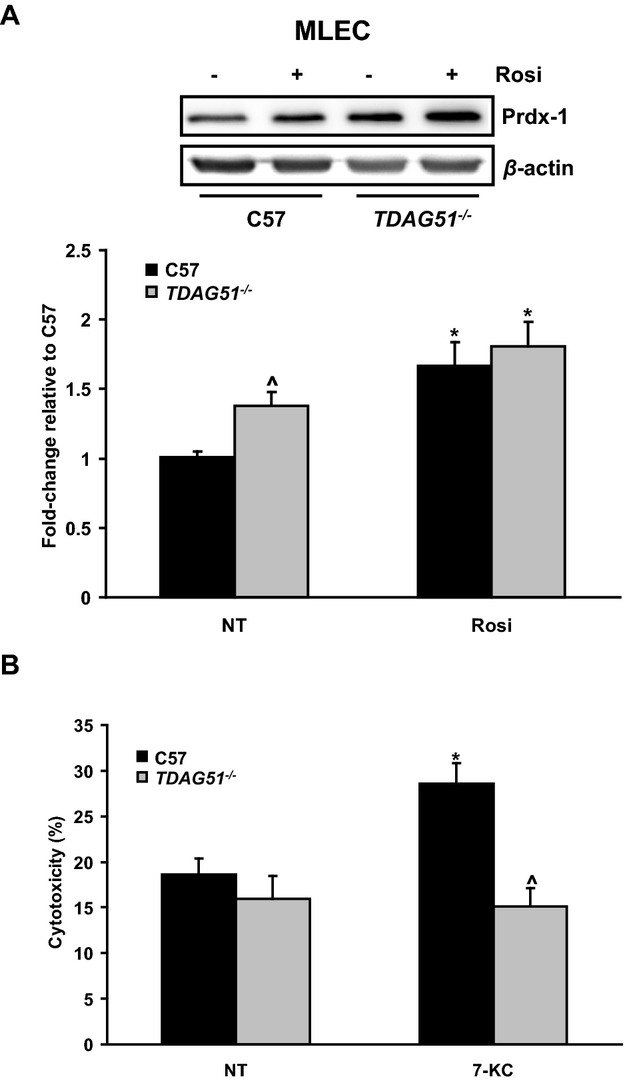
Methods and results: To study the role of TDAG51 in atherosclerosis, male mice deficient in TDAG51 and apolipoprotein E (TDAG51(-/-)/ApoE(-/-)) were generated and showed reduced atherosclerotic lesion growth (56 ± 5% reduction at 40 weeks, relative to ApoE(-/-) controls, P<0.005) and necrosis (41 ± 4% versus 63 ± 8% lesion area in TDAG51(-/-)/ApoE(-/-) and ApoE(-/-), respectively; P<0.05) without changes in plasma levels of lipids, glucose, and inflammatory cytokines. TDAG51 deficiency caused several phenotypic changes in macrophages and endothelial cells that increase cytoprotection against oxidative and ER stress, enhance PPARγ-dependent reverse cholesterol transport, and upregulate peroxiredoxin-1 (Prdx-1), an antioxidant enzyme with antiatherogenic properties (1.8 ± 0.1-fold increase in Prdx-1 protein expression, relative to control macrophages; P<0.005). Two independent case-control studies found that a genetic variant in the human TDAG51 gene region (rs2367446) is associated with CVD (OR, 1.15; 95% CI, 1.07 to 1.24; P=0.0003).
Conclusions: These findings provide evidence that TDAG51 affects specific cellular pathways known to reduce atherogenesis, suggesting that modulation of TDAG51 expression or its activity may have therapeutic benefit for the treatment of CVD.
Keywords: apoptosis; arteriosclerosis; atherosclerosis; cardiovascular diseases.
Figures
Similar articles
-
TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the cevelopment of atherosclerosis in hyperhomocysteinemia.J Biol Chem. 2003 Aug 8;278(32):30317-27. doi: 10.1074/jbc.M212897200. Epub 2003 May 8. J Biol Chem. 2003. PMID: 12738777
-
TDAG51 deficiency promotes oxidative stress-induced apoptosis through the generation of reactive oxygen species in mouse embryonic fibroblasts.Exp Mol Med. 2013 Aug 9;45(8):e35. doi: 10.1038/emm.2013.67. Exp Mol Med. 2013. PMID: 23928855 Free PMC article.
-
Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33.Cell Physiol Biochem. 2018;48(5):1995-2010. doi: 10.1159/000492522. Epub 2018 Aug 9. Cell Physiol Biochem. 2018. PMID: 30092598
-
Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux.Autophagy. 2018;14(1):120-133. doi: 10.1080/15548627.2017.1327942. Epub 2017 Nov 25. Autophagy. 2018. PMID: 28605287 Free PMC article.
-
Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E-deficient mice lacking macrophage-derived ACAT1.Circulation. 2005 May 10;111(18):2373-81. doi: 10.1161/01.CIR.0000164236.19860.13. Epub 2005 Apr 25. Circulation. 2005. PMID: 15851589
Cited by
-
T-Cell Death Associated Gene 51 Is a Novel Negative Regulator of PPARγ That Inhibits PPARγ-RXRα Heterodimer Formation in Adipogenesis.Mol Cells. 2021 Jan 31;44(1):1-12. doi: 10.14348/molcells.2020.0143. Mol Cells. 2021. PMID: 33335079 Free PMC article.
-
Multiomics Analysis of the PHLDA Gene Family in Different Cancers and Their Clinical Prognostic Value.Curr Issues Mol Biol. 2024 May 30;46(6):5488-5510. doi: 10.3390/cimb46060328. Curr Issues Mol Biol. 2024. PMID: 38921000 Free PMC article.
-
TDAG51 deficiency attenuates dextran sulfate sodium-induced colitis in mice.Sci Rep. 2022 Nov 30;12(1):20619. doi: 10.1038/s41598-022-24873-4. Sci Rep. 2022. PMID: 36450854 Free PMC article.
-
PHLDA1 Suppresses TLR4-Triggered Proinflammatory Cytokine Production by Interaction With Tollip.Front Immunol. 2022 Feb 14;13:731500. doi: 10.3389/fimmu.2022.731500. eCollection 2022. Front Immunol. 2022. PMID: 35237256 Free PMC article.
-
Transcription Factor MAFF (MAF Basic Leucine Zipper Transcription Factor F) Regulates an Atherosclerosis Relevant Network Connecting Inflammation and Cholesterol Metabolism.Circulation. 2021 May 4;143(18):1809-1823. doi: 10.1161/CIRCULATIONAHA.120.050186. Epub 2021 Feb 25. Circulation. 2021. PMID: 33626882 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
