

Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart
- PMID: 23686371
- PMCID: PMC3698799
- DOI: 10.1161/JAHA.113.004796
Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart
Abstract
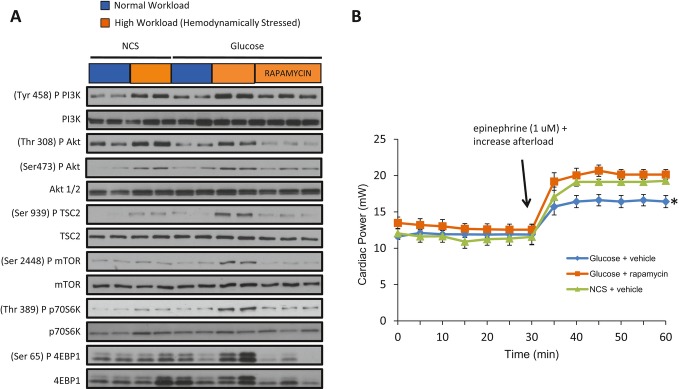
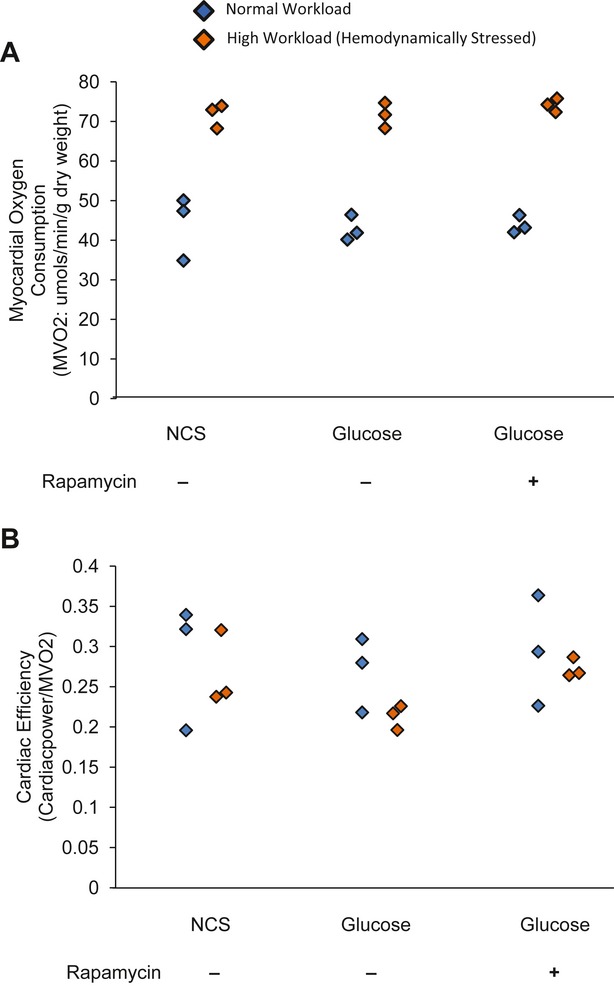
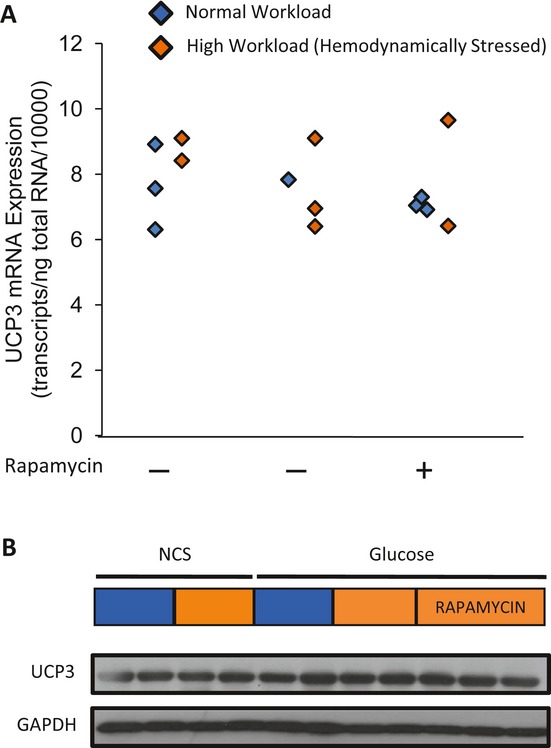
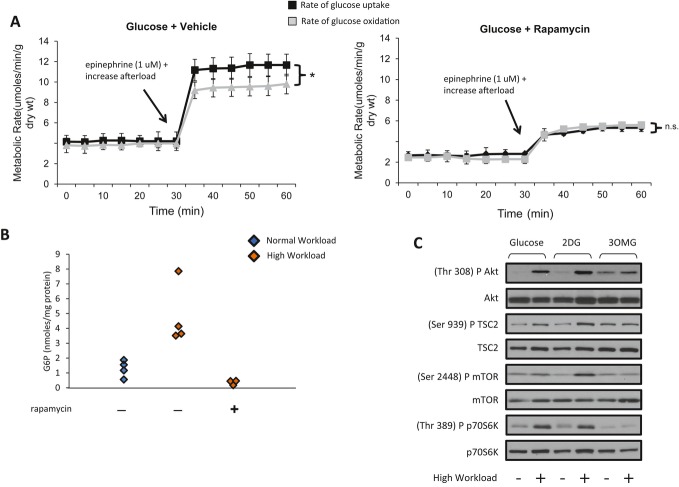
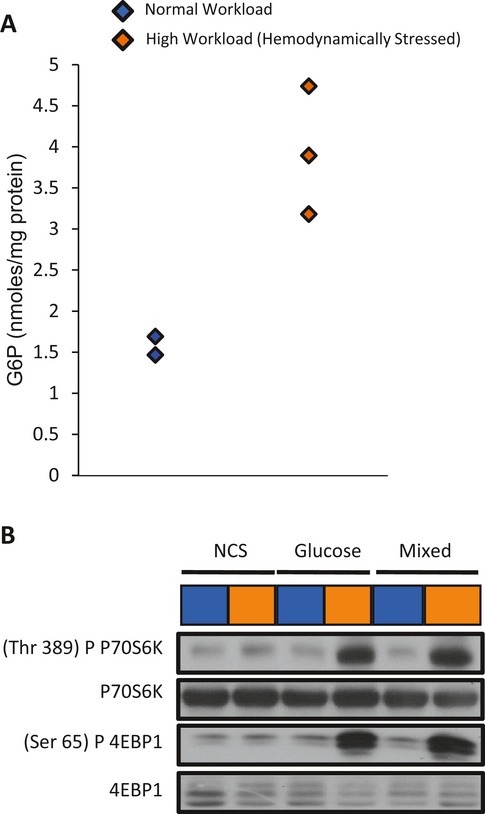
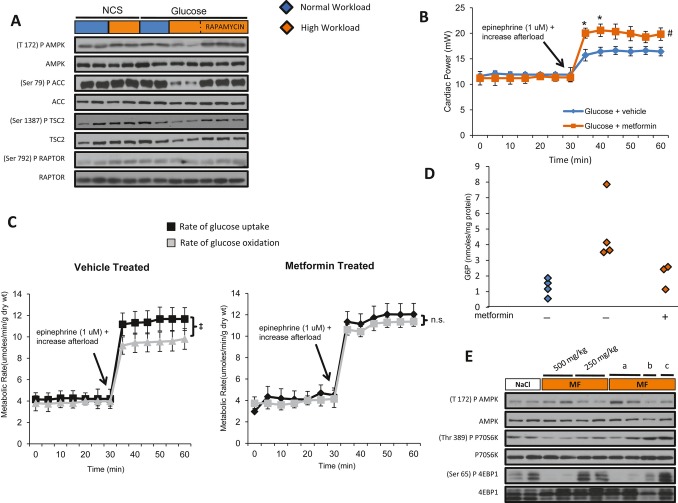
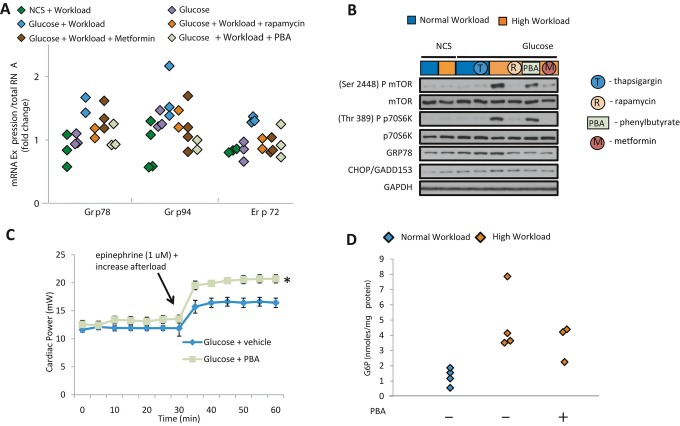
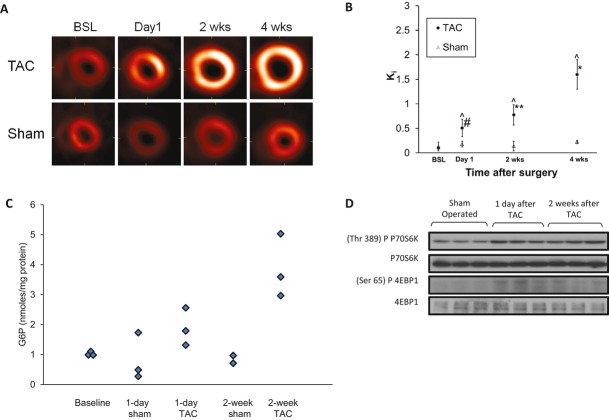
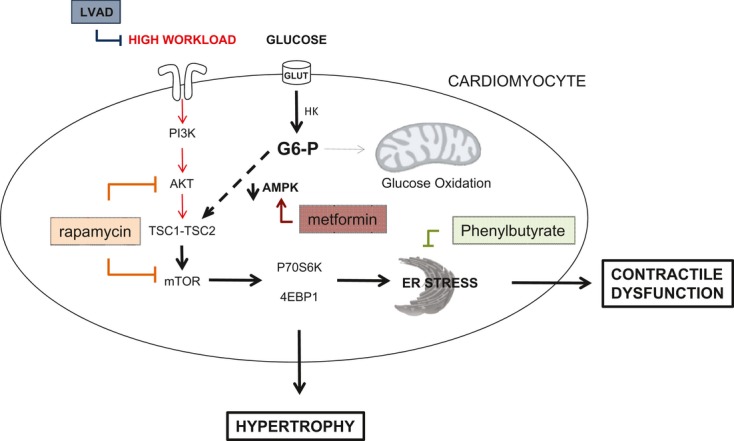
Background: Changes in energy substrate metabolism are first responders to hemodynamic stress in the heart. We have previously shown that hexose-6-phosphate levels regulate mammalian target of rapamycin (mTOR) activation in response to insulin. We now tested the hypothesis that inotropic stimulation and increased afterload also regulate mTOR activation via glucose 6-phosphate (G6P) accumulation.
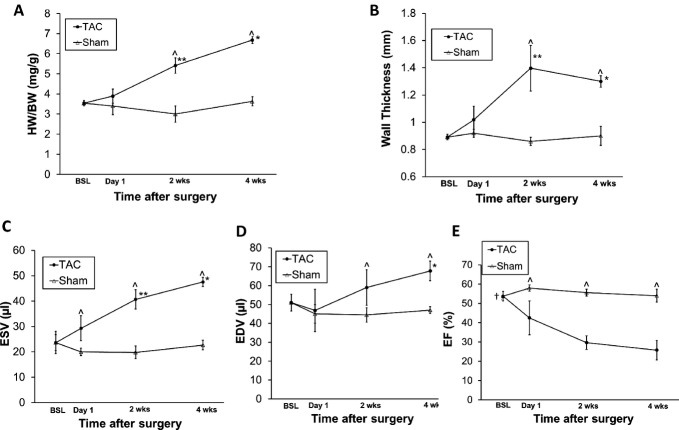
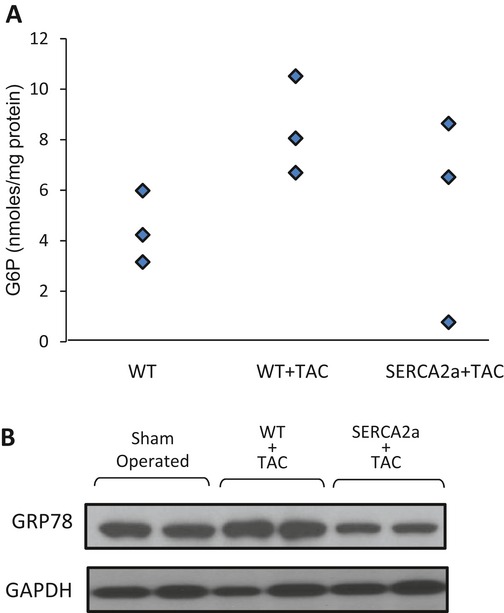
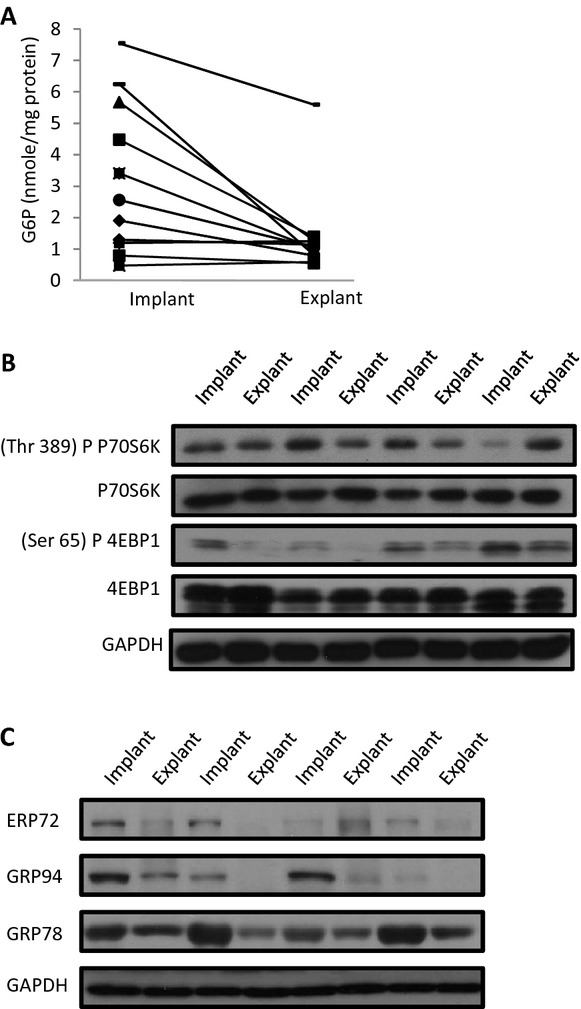
Methods and results: We subjected the working rat heart ex vivo to a high workload in the presence of different energy-providing substrates including glucose, glucose analogues, and noncarbohydrate substrates. We observed an association between G6P accumulation, mTOR activation, endoplasmic reticulum (ER) stress, and impaired contractile function, all of which were prevented by pretreating animals with rapamycin (mTOR inhibition) or metformin (AMPK activation). The histone deacetylase inhibitor 4-phenylbutyrate, which relieves ER stress, also improved contractile function. In contrast, adding the glucose analogue 2-deoxy-d-glucose, which is phosphorylated but not further metabolized, to the perfusate resulted in mTOR activation and contractile dysfunction. Next we tested our hypothesis in vivo by transverse aortic constriction in mice. Using a micro-PET system, we observed enhanced glucose tracer analog uptake and contractile dysfunction preceding dilatation of the left ventricle. In contrast, in hearts overexpressing SERCA2a, ER stress was reduced and contractile function was preserved with hypertrophy. Finally, we examined failing human hearts and found that mechanical unloading decreased G6P levels and ER stress markers.
Conclusions: We propose that glucose metabolic changes precede and regulate functional (and possibly also structural) remodeling of the heart. We implicate a critical role for G6P in load-induced mTOR activation and ER stress.
Keywords: ER stress; glucose; hypertrophy; mTOR; metabolism.
Figures
References
-
- Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998; 273:29530-29539 - PubMed
-
- Bishop S, Altschuld R. Increased glycolytic metabolism in cardiac hypertrophy and congestive heart failure. Am J Physiol. 1970; 218:153-159 - PubMed
-
- Taegtmeyer H, Overturf ML. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension. 1988; 11:416-426 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
