

Is SOD1 loss of function involved in amyotrophic lateral sclerosis?
- PMID: 23687121
- PMCID: PMC3722346
- DOI: 10.1093/brain/awt097
Is SOD1 loss of function involved in amyotrophic lateral sclerosis?
Abstract
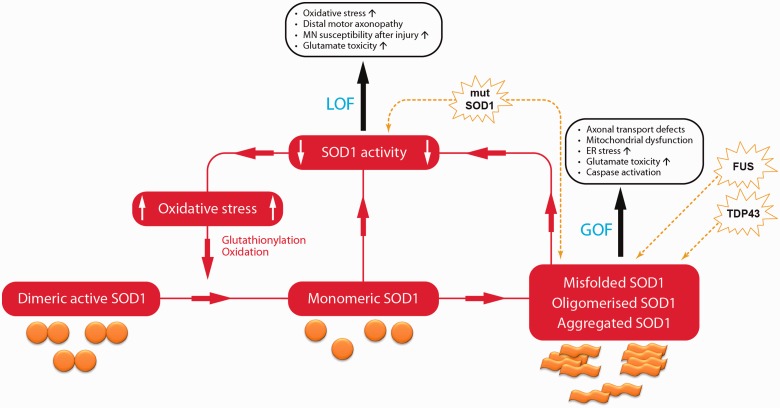
Mutations in the gene superoxide dismutase 1 (SOD1) are causative for familial forms of the neurodegenerative disease amyotrophic lateral sclerosis. When the first SOD1 mutations were identified they were postulated to give rise to amyotrophic lateral sclerosis through a loss of function mechanism, but experimental data soon showed that the disease arises from a--still unknown--toxic gain of function, and the possibility that loss of function plays a role in amyotrophic lateral sclerosis pathogenesis was abandoned. Although loss of function is not causative for amyotrophic lateral sclerosis, here we re-examine two decades of evidence regarding whether loss of function may play a modifying role in SOD1-amyotrophic lateral sclerosis. From analysing published data from patients with SOD1-amyotrophic lateral sclerosis, we find a marked loss of SOD1 enzyme activity arising from almost all mutations. We continue to examine functional data from all Sod1 knockout mice and we find obvious detrimental effects within the nervous system with, interestingly, some specificity for the motor system. Here, we bring together historical and recent experimental findings to conclude that there is a possibility that SOD1 loss of function may play a modifying role in amyotrophic lateral sclerosis. This likelihood has implications for some current therapies aimed at knocking down the level of mutant protein in patients with SOD1-amyotrophic lateral sclerosis. Finally, the wide-ranging phenotypes that result from loss of function indicate that SOD1 gene sequences should be screened in diseases other than amyotrophic lateral sclerosis.
Keywords: amyotrophic lateral sclerosis; loss of function; motor neuron disease; superoxide dismutase 1.
Figures

References
-
- Acsadi G, Lee I, Li X, Khaidakov M, Pecinova A, Parker GC, et al. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J Neurosci Res. 2009;87:2748–56. - PubMed
-
- Andersen PM, Al Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–15. - PubMed
-
- Andersen PM, Nilsson P, Ala-Hurula V, Keranen ML, Tarvainen I, Haltia T, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet. 1995;10:61–6. - PubMed
-
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71:2041–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
