

Impact of peripheral ketolytic deficiency on hepatic ketogenesis and gluconeogenesis during the transition to birth
- PMID: 23689508
- PMCID: PMC3707678
- DOI: 10.1074/jbc.M113.454868
Impact of peripheral ketolytic deficiency on hepatic ketogenesis and gluconeogenesis during the transition to birth
Abstract
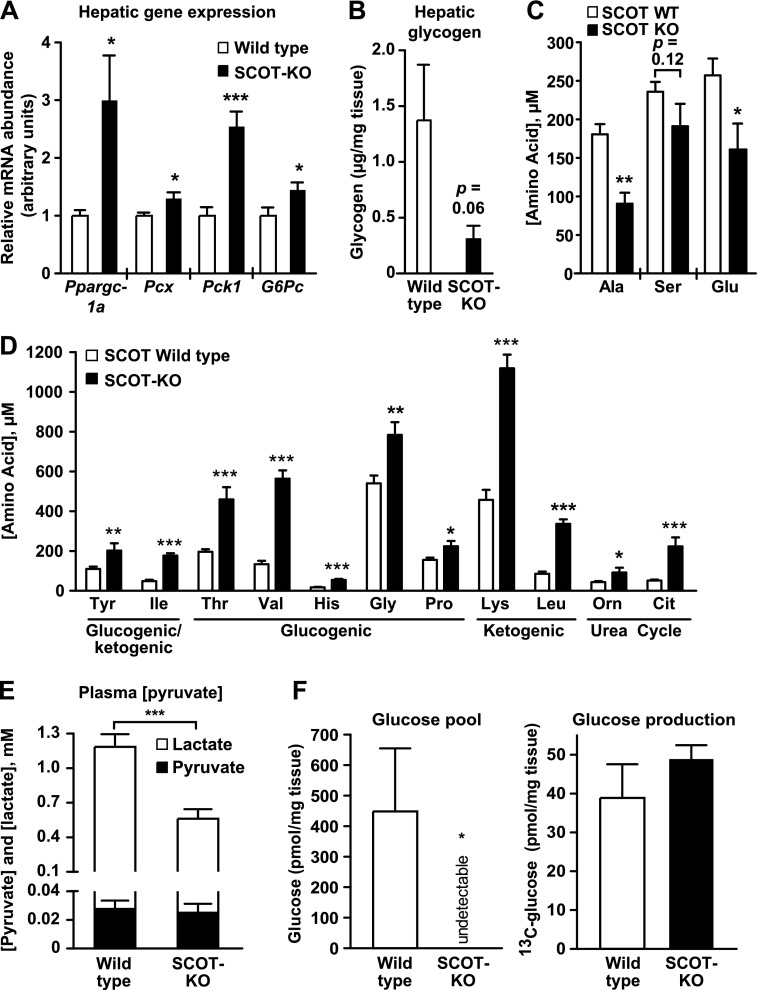
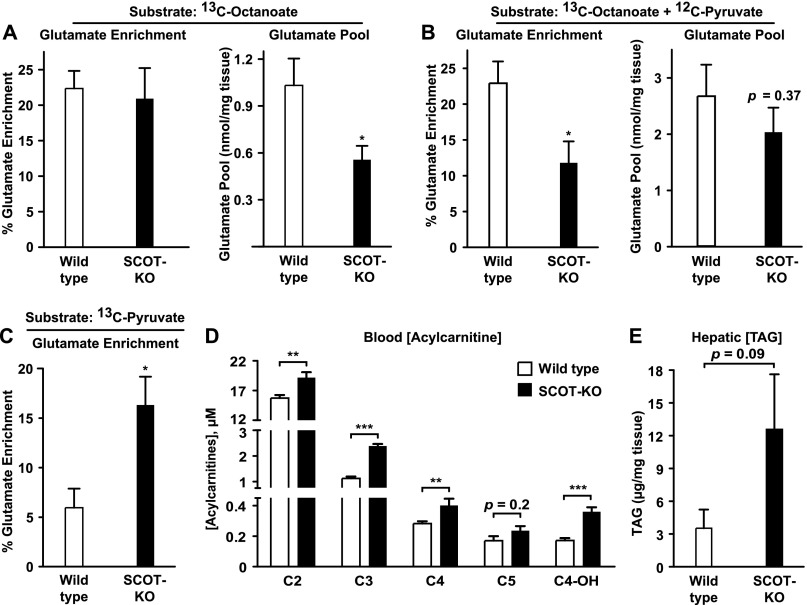
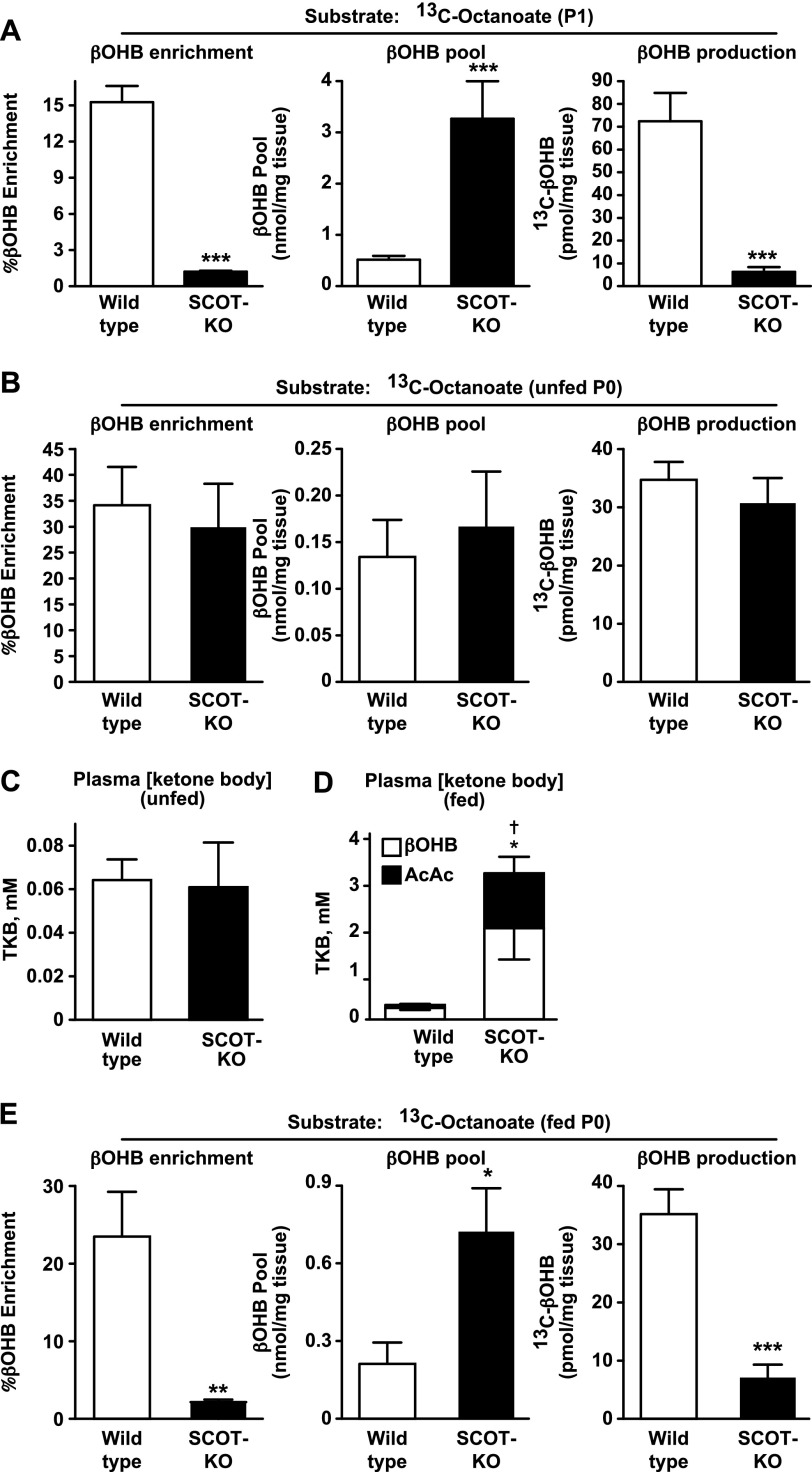
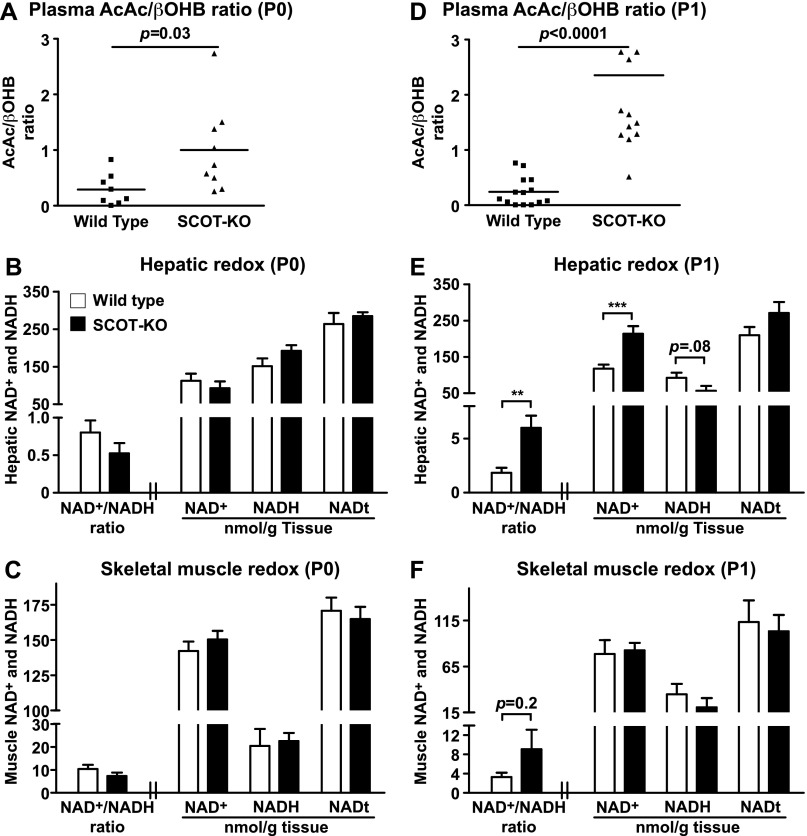
Preservation of bioenergetic homeostasis during the transition from the carbohydrate-laden fetal diet to the high fat, low carbohydrate neonatal diet requires inductions of hepatic fatty acid oxidation, gluconeogenesis, and ketogenesis. Mice with loss-of-function mutation in the extrahepatic mitochondrial enzyme CoA transferase (succinyl-CoA:3-oxoacid CoA transferase, SCOT, encoded by nuclear Oxct1) cannot terminally oxidize ketone bodies and develop lethal hyperketonemic hypoglycemia within 48 h of birth. Here we use this model to demonstrate that loss of ketone body oxidation, an exclusively extrahepatic process, disrupts hepatic intermediary metabolic homeostasis after high fat mother's milk is ingested. Livers of SCOT-knock-out (SCOT-KO) neonates induce the expression of the genes encoding peroxisome proliferator-activated receptor γ co-activator-1a (PGC-1α), phosphoenolpyruvate carboxykinase (PEPCK), pyruvate carboxylase, and glucose-6-phosphatase, and the neonate's pools of gluconeogenic alanine and lactate are each diminished by 50%. NMR-based quantitative fate mapping of (13)C-labeled substrates revealed that livers of SCOT-KO newborn mice synthesize glucose from exogenously administered pyruvate. However, the contribution of exogenous pyruvate to the tricarboxylic acid cycle as acetyl-CoA is increased in SCOT-KO livers and is associated with diminished terminal oxidation of fatty acids. After mother's milk provokes hyperketonemia, livers of SCOT-KO mice diminish de novo hepatic β-hydroxybutyrate synthesis by 90%. Disruption of β-hydroxybutyrate production increases hepatic NAD(+)/NADH ratios 3-fold, oxidizing redox potential in liver but not skeletal muscle. Together, these results indicate that peripheral ketone body oxidation prevents hypoglycemia and supports hepatic metabolic homeostasis, which is critical for the maintenance of glycemia during the adaptation to birth.
Keywords: Coenzyme A Transferase; Gluconeogenesis; Glucose Homeostasis; Ketone Body Metabolism; Liver Metabolism; NMR Substrate Fate Mapping; Neonatal Metabolism; Redox; Tricarboxylic Acid (TCA) Cycle.
Figures


References
-
- Girard J., Ferré P., Pégorier J. P., Duée P. H. (1992) Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol. Rev. 72, 507–562 - PubMed
-
- Ward Platt M., Deshpande S. (2005) Metabolic adaptation at birth. Semin. Fetal Neonatal Med. 10, 341–350 - PubMed
-
- Robinson A. M., Williamson D. H. (1980) Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 60, 143–187 - PubMed
-
- McGarry J. D., Foster D. W. (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 49, 395–420 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
