

Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2
- PMID: 23692823
- PMCID: PMC3670812
- DOI: 10.1186/1750-1172-8-80
Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2
Abstract
Background: Early onset epileptic encephalopathies (EOEEs) are dramatic heterogeneous conditions in which aetiology, seizures and/or interictal EEG have a negative impact on neurological development. Several genes have been associated with EOEE and a molecular diagnosis workup is challenging since similar phenotypes are associated with mutations in different genes and since mutations in one given gene can be associated with very different phenotypes. Recently, de novo mutations in KCNQ2, have been found in about 10% of EOEE patients. Our objective was to confirm that KCNQ2 was an important gene to include in the diagnosis workup of EOEEs and to fully describe the clinical and EEG features of mutated patients.
Methods: We have screened KCNQ2 in a cohort of 71 patients with an EOEE, without any brain structural abnormality. To be included in the cohort, patient's epilepsy should begin before three months of age and be associated with abnormal interictal EEG and neurological impairment. Brain MRI should not show any structural abnormality that could account for the epilepsy.
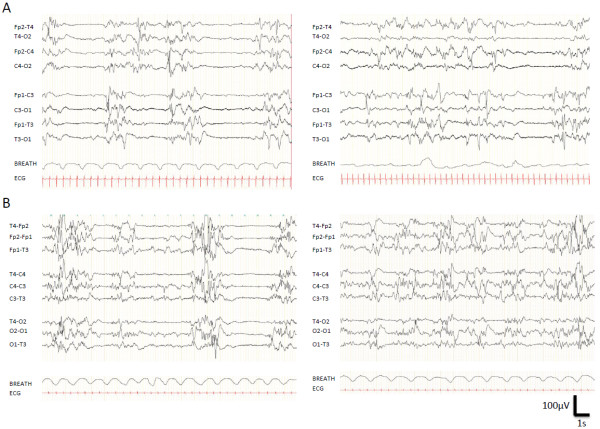
Results: Out of those 71 patients, 16 had a de novo mutation in KCNQ2 (23%). Interestingly, in the majority of the cases, the initial epileptic features of these patients were comparable to those previously described in the case of benign familial neonatal epilepsy (BFNE) also caused by KCNQ2 mutations. However, in contrast to BFNE, the interictal background EEG was altered and displayed multifocal spikes or a suppression-burst pattern. The ongoing epilepsy and development were highly variable but overall severe: 15/16 had obvious cognitive impairment, half of the patients became seizure-free, 5/16 could walk before the age of 3 and only 2/16 patient acquired the ability to speak.
Conclusion: This study confirms that KCNQ2 is frequently mutated de novo in neonatal onset epileptic encephalopathy. We show here that despite a relatively stereotyped beginning of the condition, the neurological and epileptic evolution is variable.
Figures
References
-
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. - DOI - PubMed
-
- Plouin P. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven; 1997. Benign familial neonatal convulsions and benign idiopathic neonatal convulsions; pp. 2247–2249.
-
- Bellini G, Miceli F, Soldovieri MV, Miraglia Del Giudice E, Coppola G, Taglialatela M. In: GeneReviews™ [Internet] Pagon RA, Bird TD, Dolan CR, editor. Seattle (WA): University of Washington, Seattle: ; 1993. KCNQ2 related disorders. 2010 Apr 27 [Updated 2013 Apr 11] Available from: http://www.ncbi.nlm.nih.gov/books/NBK32534/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
