

HMGB1 accelerates alveolar epithelial repair via an IL-1β- and αvβ6 integrin-dependent activation of TGF-β1
- PMID: 23696858
- PMCID: PMC3655948
- DOI: 10.1371/journal.pone.0063907
HMGB1 accelerates alveolar epithelial repair via an IL-1β- and αvβ6 integrin-dependent activation of TGF-β1
Erratum in
- PLoS One. 2013;8(10). doi:10.1371/annotation/88f820f2-18dd-4d3b-8989-68f170b26b04
Abstract
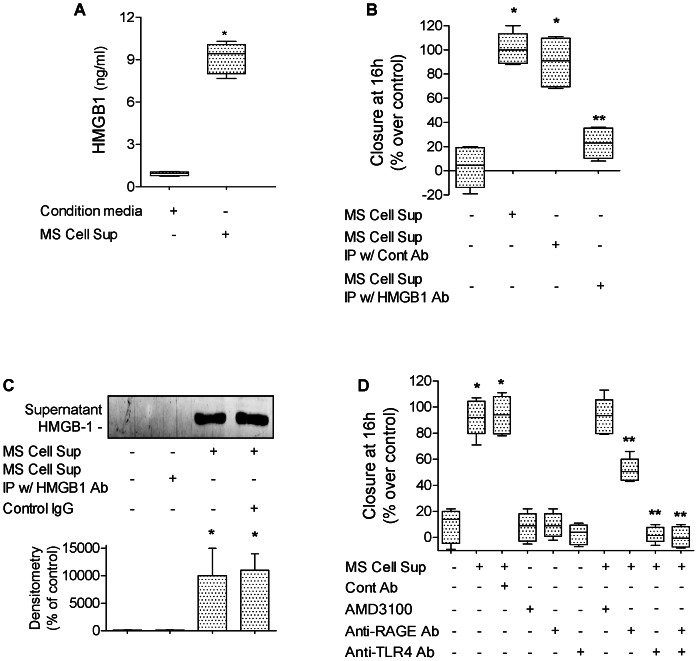
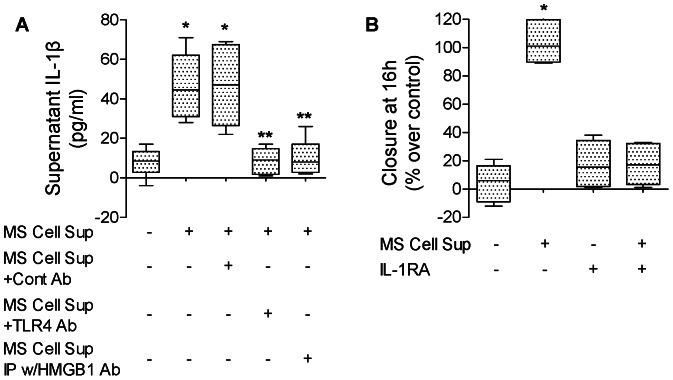
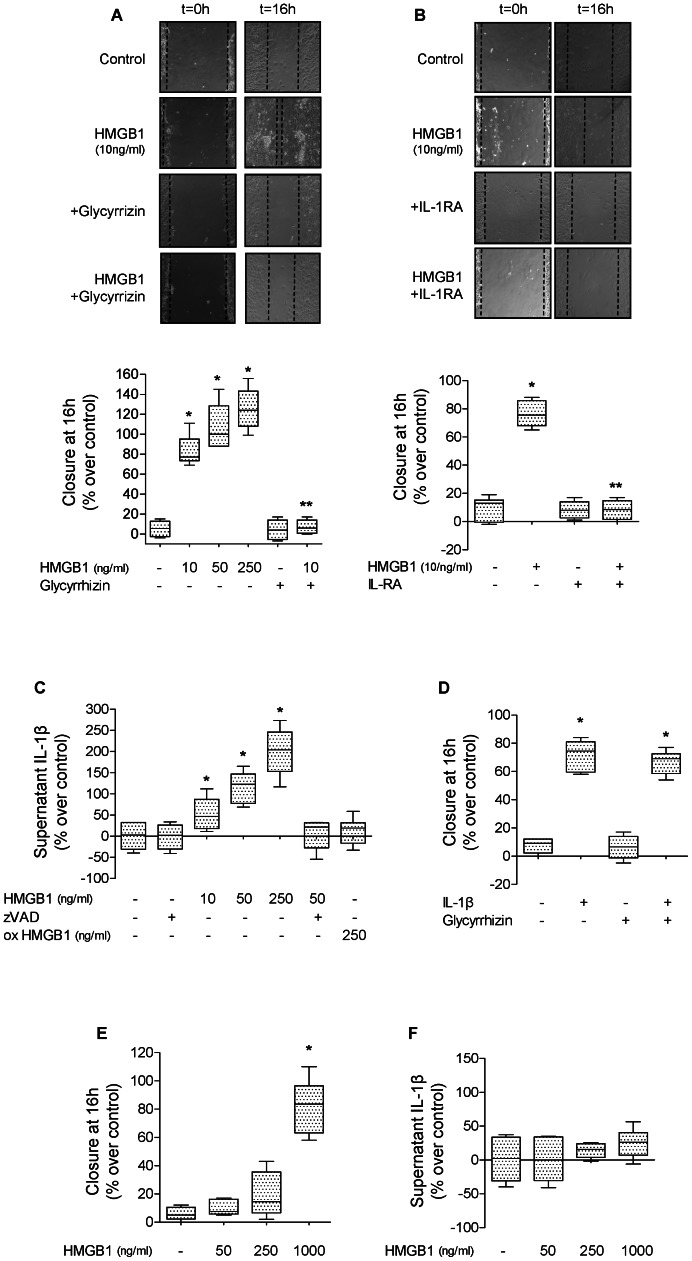
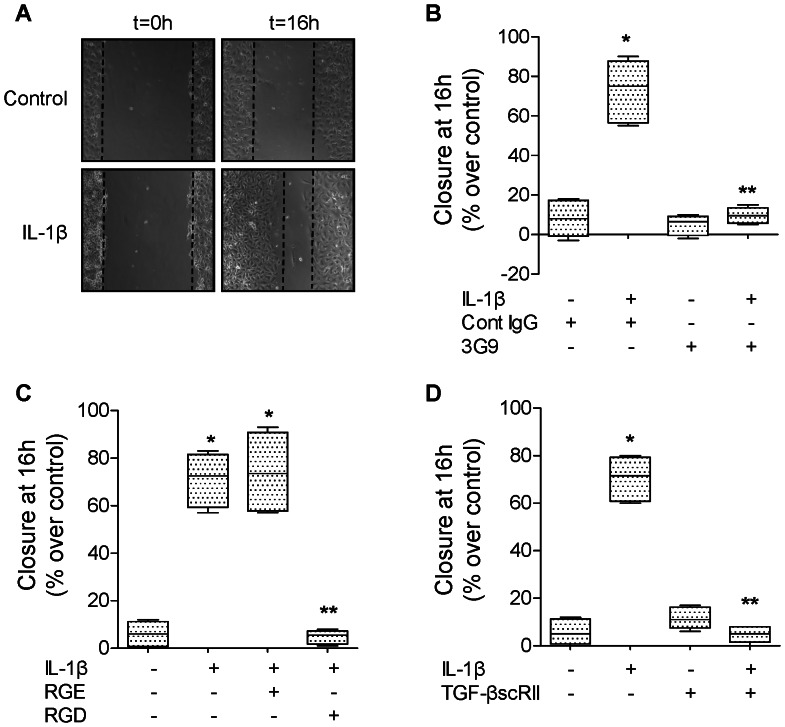
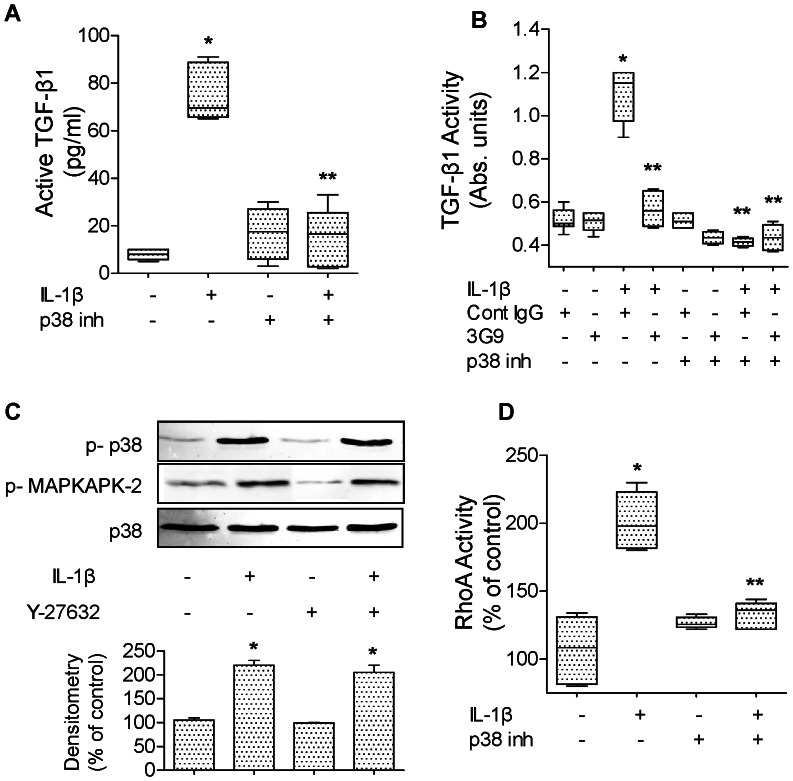
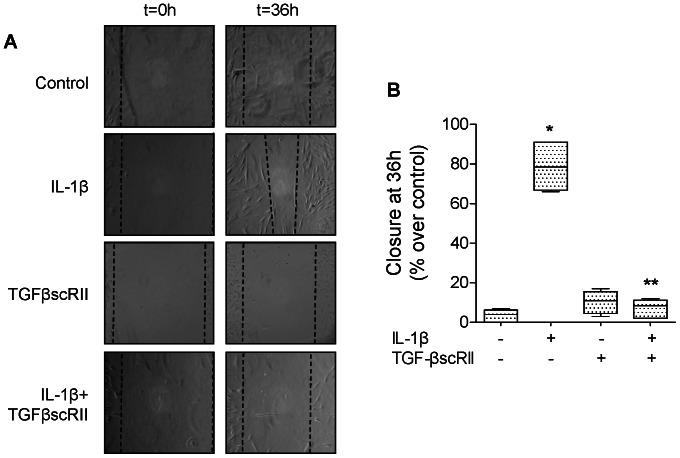
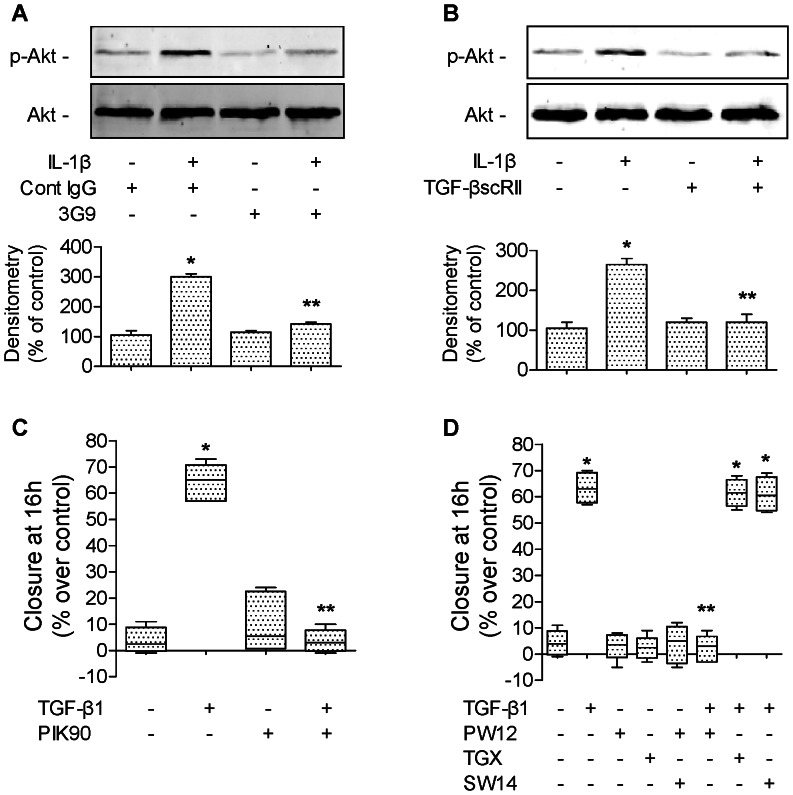
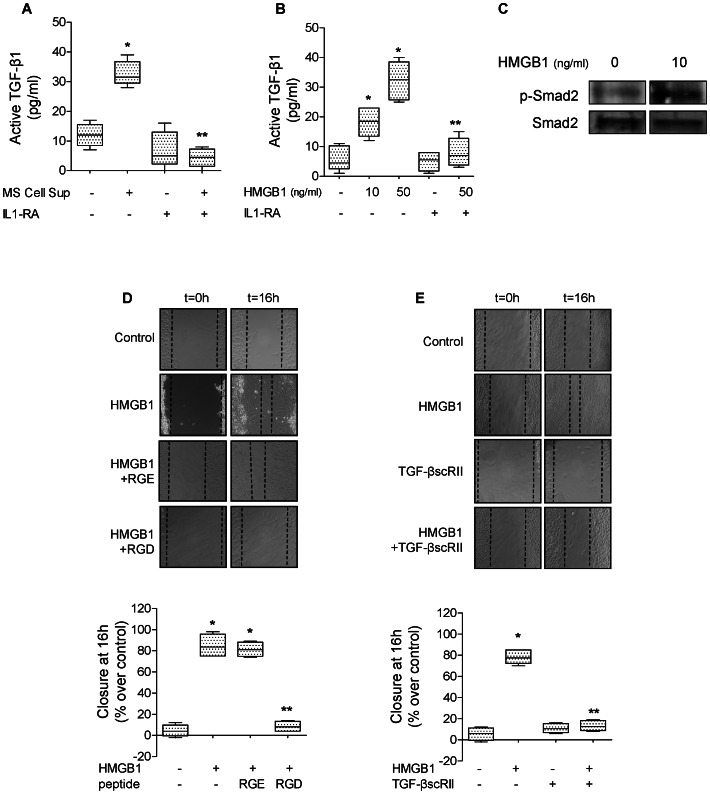
High mobility group box 1 (HMGB1) protein is a danger-signaling molecule, known to activate an inflammatory response via TLR4 and RAGE. HMGB1 can be either actively secreted or passively released from damaged alveolar epithelial cells. Previous studies have shown that IL-1β, a critical mediator acute lung injury in humans that is activated by HMGB1, enhances alveolar epithelial repair, although the mechanisms are not fully understood. Herein, we tested the hypothesis that HMGB1 released by wounded alveolar epithelial cells would increase primary rat and human alveolar type II cell monolayer wound repair via an IL-1β-dependent activation of TGF-β1. HMGB1 induced in primary cultures of rat alveolar epithelial cells results in the release of IL-1β that caused the activation of TGF-β1 via a p38 MAPK-, RhoA- and αvβ6 integrin-dependent mechanism. Furthermore, active TGF-β1 accelerated the wound closure of primary rat epithelial cell monolayers via a PI3 kinase α-dependent mechanism. In conclusion, this study demonstrates that HMGB1 released by wounded epithelial cell monolayers, accelerates wound closure in the distal lung epithelium via the IL-1β-mediated αvβ6-dependent activation of TGF-β1, and thus could play an important role in the resolution of acute lung injury by promoting repair of the injured alveolar epithelium.
Conflict of interest statement
Figures
References
-
- Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349. - PubMed
-
- Geiser T, Jarreau PH, Atabai K, Matthay MA (2000) Interleukin-1beta augments in vitro alveolar epithelial repair. Am J Physiol Lung Cell Mol Physiol 279: L1184–1190. - PubMed
-
- Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, et al. (2001) Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med 163: 1384–1388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
