

Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing
- PMID: 23698529
- PMCID: PMC3719633
- DOI: 10.1128/JCM.00066-13
Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing
Abstract
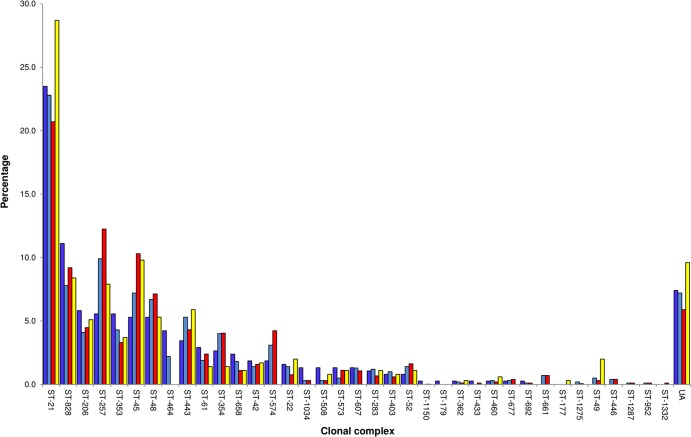
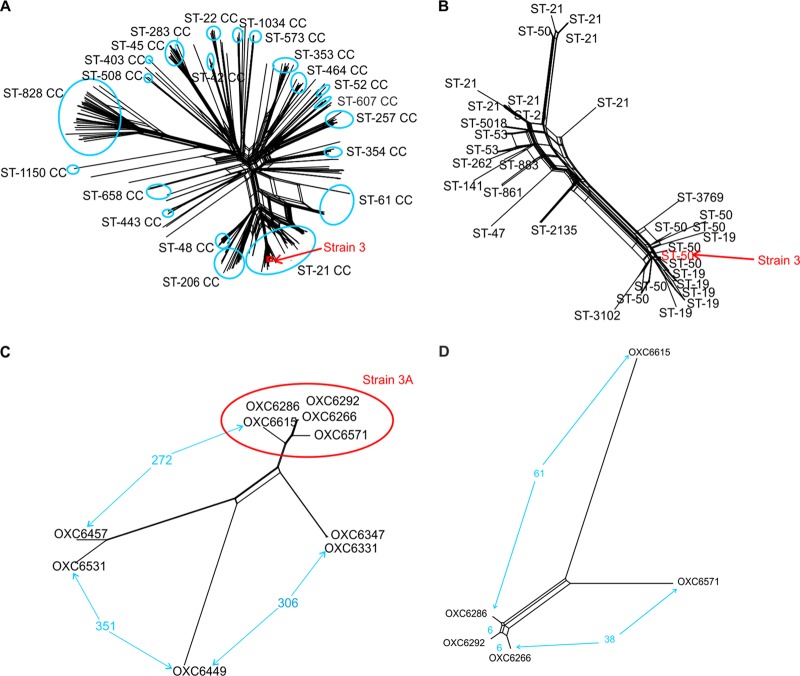
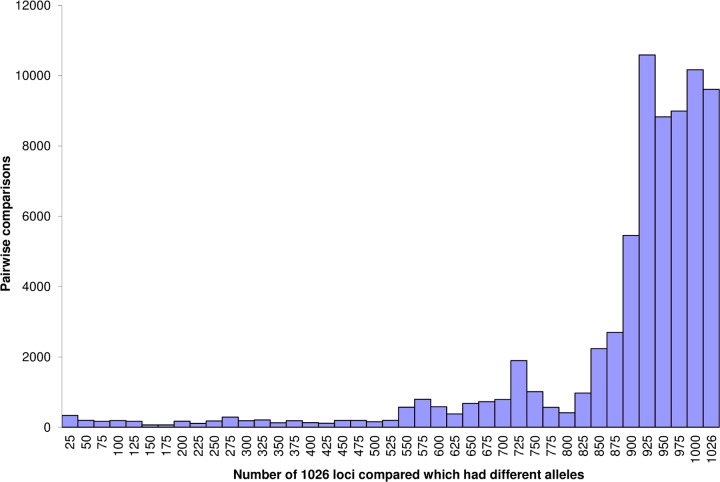
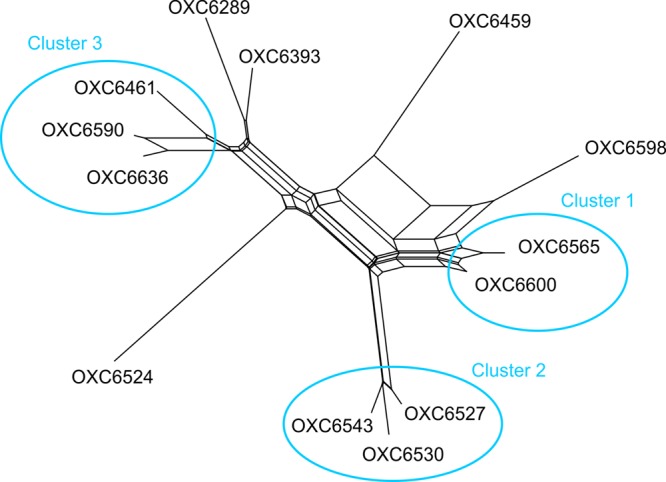
Sequence-based typing is essential for understanding the epidemiology of Campylobacter infections, a major worldwide cause of bacterial gastroenteritis. We demonstrate the practical and rapid exploitation of whole-genome sequencing to provide routine definitive characterization of Campylobacter jejuni and Campylobacter coli for clinical and public health purposes. Short-read data from 384 Campylobacter clinical isolates collected over 4 months in Oxford, United Kingdom, were assembled de novo. Contigs were deposited at the pubMLST.org/campylobacter website and automatically annotated for 1,667 loci. Typing and phylogenetic information was extracted and comparative analyses were performed for various subsets of loci, up to the level of the whole genome, using the Genome Comparator and Neighbor-net algorithms. The assembled sequences (for 379 isolates) were diverse and resembled collections from previous studies of human campylobacteriosis. Small subsets of very closely related isolates originated mainly from repeated sampling from the same patients and, in one case, likely laboratory contamination. Much of the within-patient variation occurred in phase-variable genes. Clinically and epidemiologically informative data can be extracted from whole-genome sequence data in real time with straightforward, publicly available tools. These analyses are highly scalable, are transparent, do not require closely related genome reference sequences, and provide improved resolution (i) among Campylobacter clonal complexes and (ii) between very closely related isolates. Additionally, these analyses rapidly differentiated unrelated isolates, allowing the detection of single-strain clusters. The approach is widely applicable to analyses of human bacterial pathogens in real time in clinical laboratories, with little specialist training required.
Figures
References
-
- Fournier P-E, Drancourt M, Raoult D. 2007. Bacterial genome sequencing and its use in infectious diseases. Lancet Infect. Dis. 7:711–723 - PubMed
-
- Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, Sanders M, Enright MC, Dougan G, Bentley SD, Parkhill J, Fraser LJ, Betley JR, Schulz-Trieglaff OB, Smith GP, Peacock SJ. 2012. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 366:2267–2275 - PMC - PubMed
-
- Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 - PMC - PubMed
-
- Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, Ip CL, Wilson DJ, Didelot X, O'Connor L, Lay R, Buck D, Kearns AM, Shaw A, Paul J, Wilcox MH, Donnelly PJ, Peto TE, Walker AS, Crook DW. 2012. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2:e001124. 10.1136/bmjopen-2012-001124 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
