

Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma
- PMID: 23699600
- PMCID: PMC3709654
- DOI: 10.1182/blood-2013-03-487884
Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma
Abstract
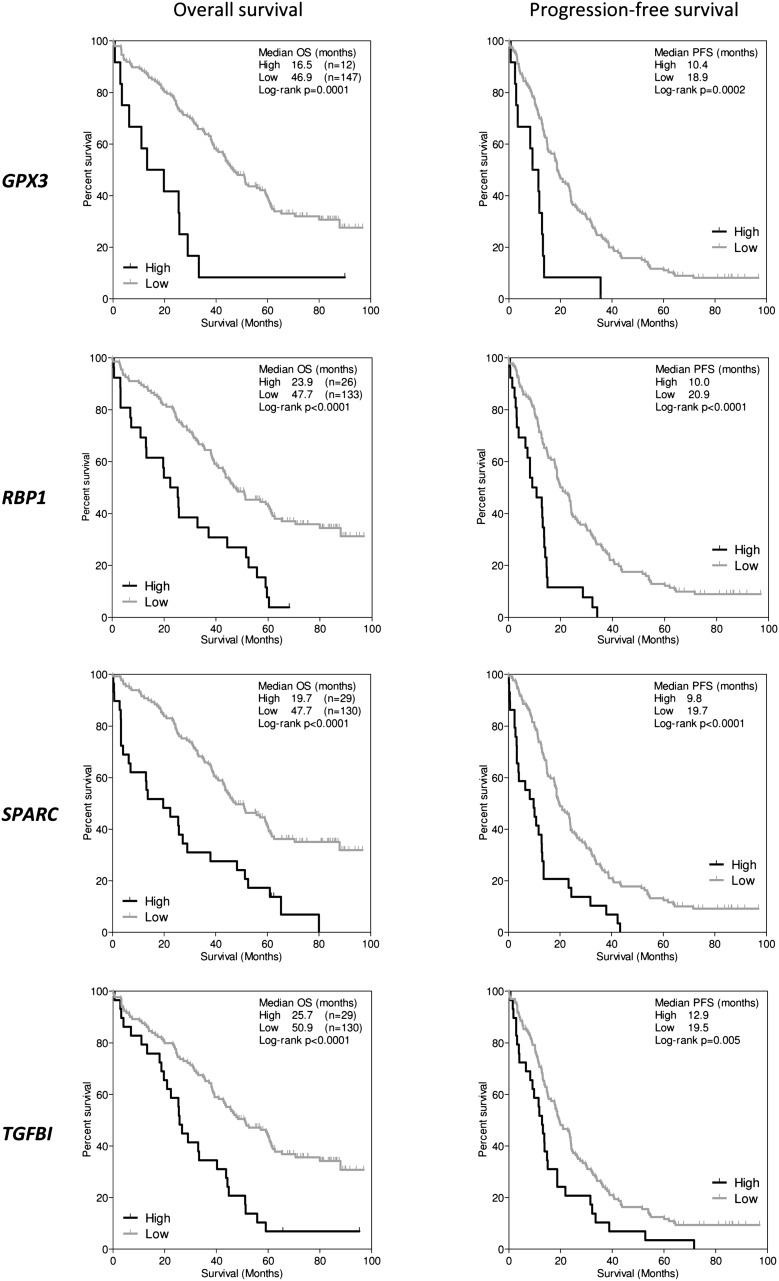
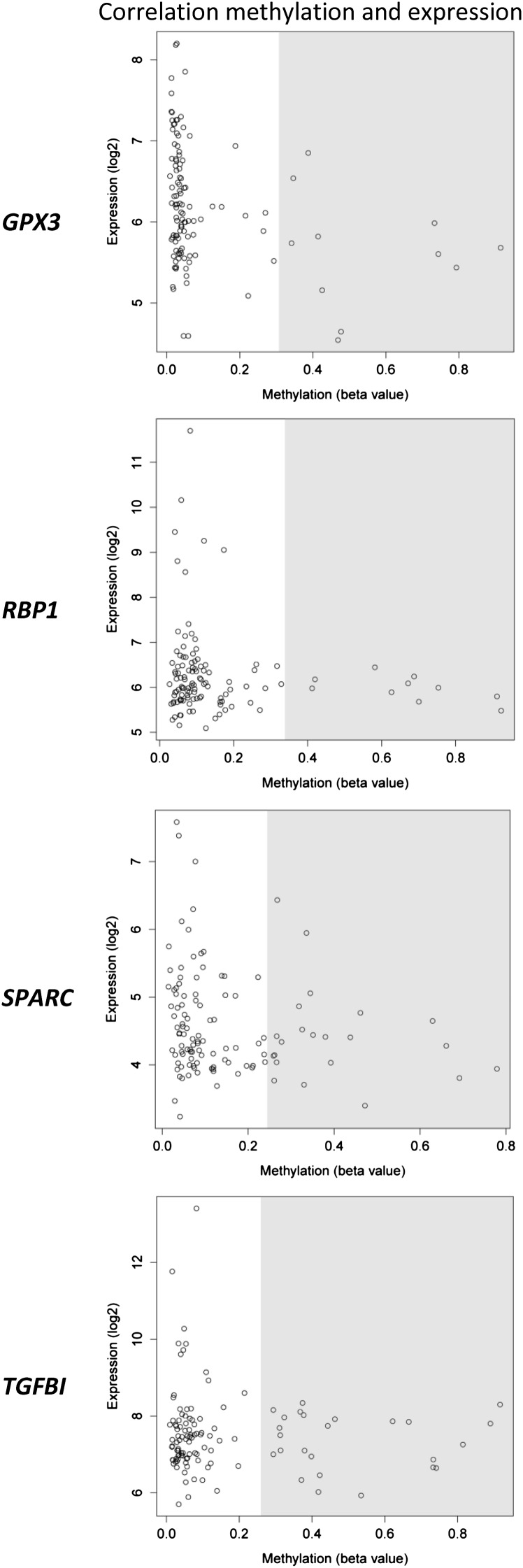
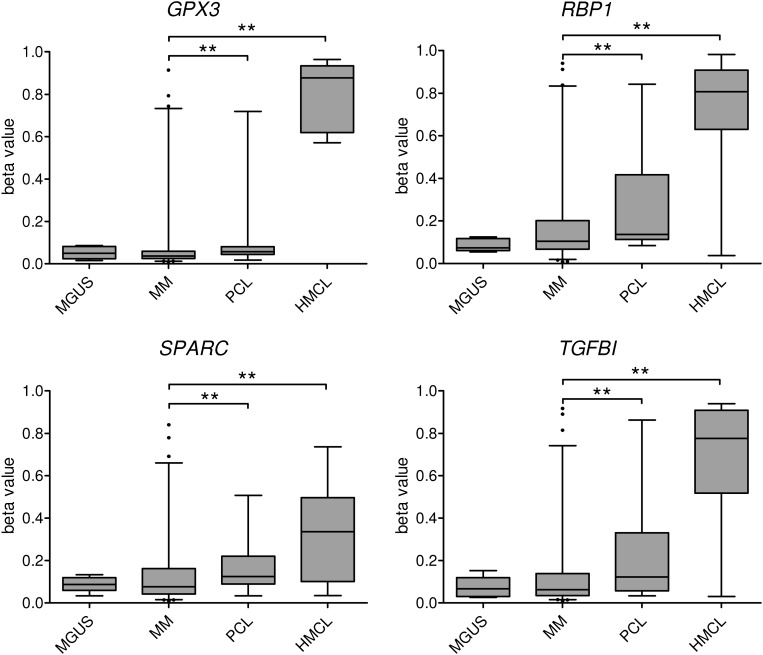
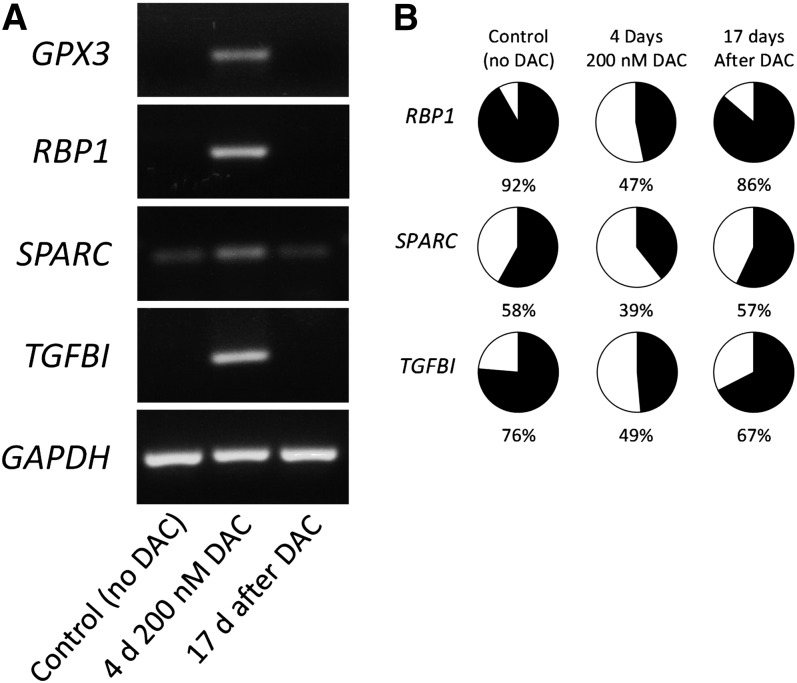
Outcome in multiple myeloma is highly variable and a better understanding of the factors that influence disease biology is essential to understand and predict behavior in individual patients. In the present study, we analyzed combined genomewide DNA methylation and gene expression data of patients treated in the Medical Research Council Myeloma IX trial. We used these data to identify epigenetically repressed tumor suppressor genes with prognostic relevance in myeloma. We identified 195 genes with changes in methylation status that were significantly associated with prognosis. Combining DNA methylation and gene expression data led to the identification of the epigenetically regulated tumor modulating genes GPX3, RBP1, SPARC, and TGFBI. Hypermethylation of these genes was associated with significantly shorter overall survival, independent of age, International Staging System score, and adverse cytogenetics. The 4 differentially methylated and expressed genes are known to mediate important tumor suppressive functions including response to chemotherapy (TGFBI), interaction with the microenvironment (SPARC), retinoic acid signaling (RBP1), and the response to oxidative stress (GPX3), which could explain the prognostic impact of their differential methylation. Assessment of the DNA methylation status of the identified genes could contribute to the molecular characterization of myeloma, which is prerequisite for an individualized treatment approach.
Figures
References
-
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. - PubMed
-
- Walker BA, Wardell CP, Chiecchio L, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117(2):553–562. - PubMed
-
- Deneberg S, Guardiola P, Lennartsson A, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood. 2011;118(20):5573–5582. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
