

Identification and characterisation of mutations associated with von Willebrand disease in a Turkish patient cohort
- PMID: 23702511
- PMCID: PMC4213552
- DOI: 10.1160/TH13-02-0135
Identification and characterisation of mutations associated with von Willebrand disease in a Turkish patient cohort
Abstract
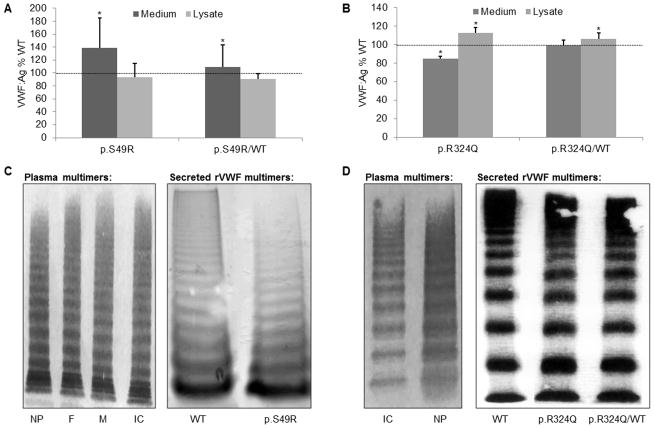
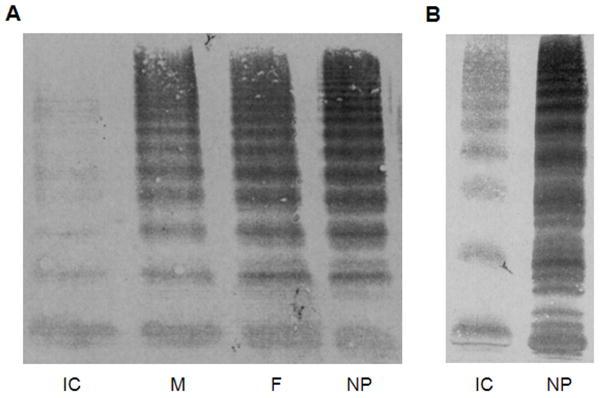
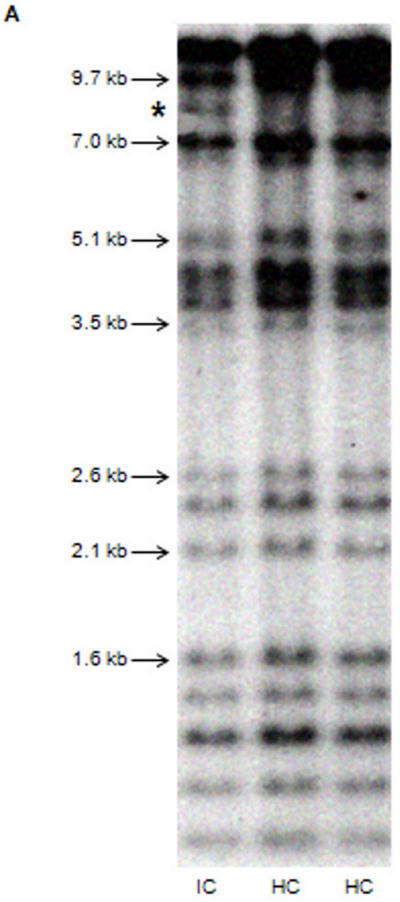
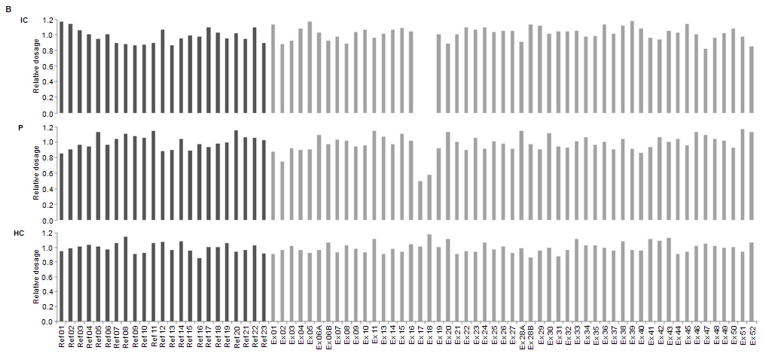
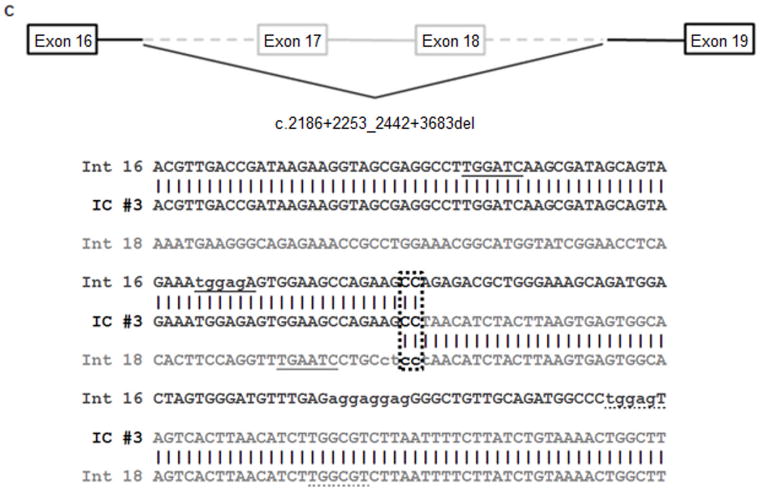
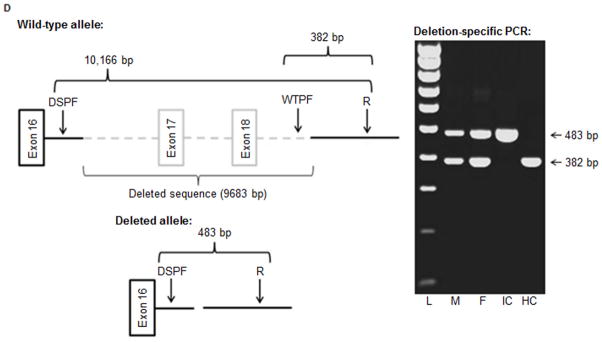
Several cohort studies have investigated the molecular basis of von Willebrand disease (VWD); however, these have mostly focused on European and North American populations. This study aimed to investigate mutation spectrum in 26 index cases (IC) from Turkey diagnosed with all three VWD types, the majority (73%) with parents who were knowingly related. IC were screened for mutations using multiplex ligation-dependent probe amplification and analysis of all von Willebrand factor gene (VWF) exons and exon/intron boundaries. Selected missense mutations were expressed in vitro. Candidate VWF mutations were identified in 25 of 26 IC and included propeptide missense mutations in four IC (two resulting in type 1 and two in recessive 2A), all influencing VWF expression in vitro. Four missense mutations, a nonsense mutation and a small in-frame insertion resulting in type 2A were also identified. Of 15 type 3 VWD IC, 13 were homozygous and two compound heterozygous for 14 candidate mutations predicted to result in lack of expression and two propeptide missense changes. Identification of intronic breakpoints of an exon 17-18 deletion suggested that the mutation resulted from non-homologous end joining. This study provides further insight into the pathogenesis of VWD in a population with a high degree of consanguineous partnerships.
Conflict of interest statement
None declared.
Figures
References
-
- Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. - PubMed
-
- Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69:454–9. - PubMed
-
- Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123:893–8. - PubMed
-
- Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101:2089–93. - PubMed
-
- Gupta PK, Ahmed RPH, Sazawal S, et al. Relatively high frequency of VWD types 3 and 2 in a cohort of Indian patients: the role of multimeric analysis. J Thromb Haemost. 2005;3:1321–2. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
