

Combined sequence-based and genetic mapping analysis of complex traits in outbred rats
- PMID: 23708188
- PMCID: PMC3821058
- DOI: 10.1038/ng.2644
Combined sequence-based and genetic mapping analysis of complex traits in outbred rats
Abstract
Genetic mapping on fully sequenced individuals is transforming understanding of the relationship between molecular variation and variation in complex traits. Here we report a combined sequence and genetic mapping analysis in outbred rats that maps 355 quantitative trait loci for 122 phenotypes. We identify 35 causal genes involved in 31 phenotypes, implicating new genes in models of anxiety, heart disease and multiple sclerosis. The relationship between sequence and genetic variation is unexpectedly complex: at approximately 40% of quantitative trait loci, a single sequence variant cannot account for the phenotypic effect. Using comparable sequence and mapping data from mice, we show that the extent and spatial pattern of variation in inbred rats differ substantially from those of inbred mice and that the genetic variants in orthologous genes rarely contribute to the same phenotype in both species.
Figures
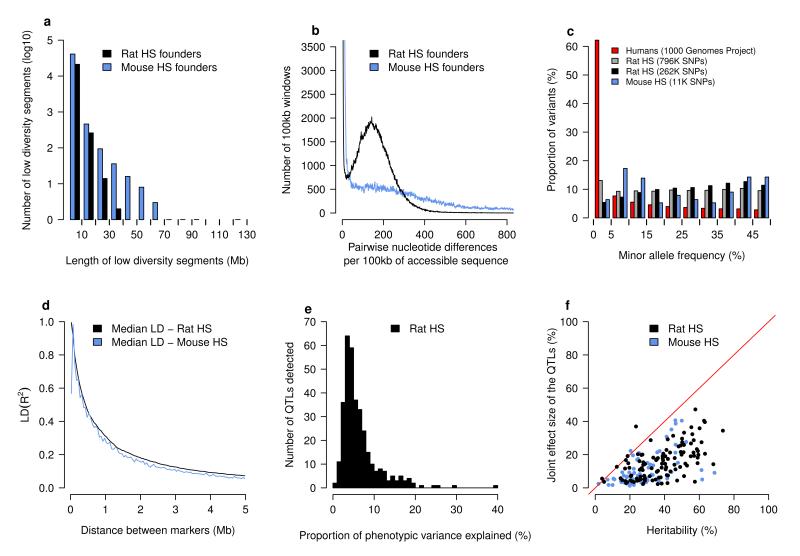
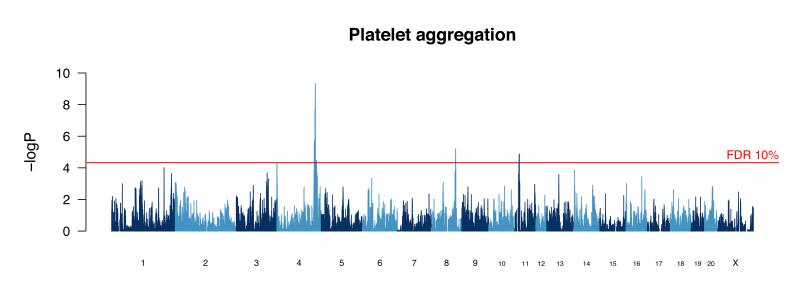
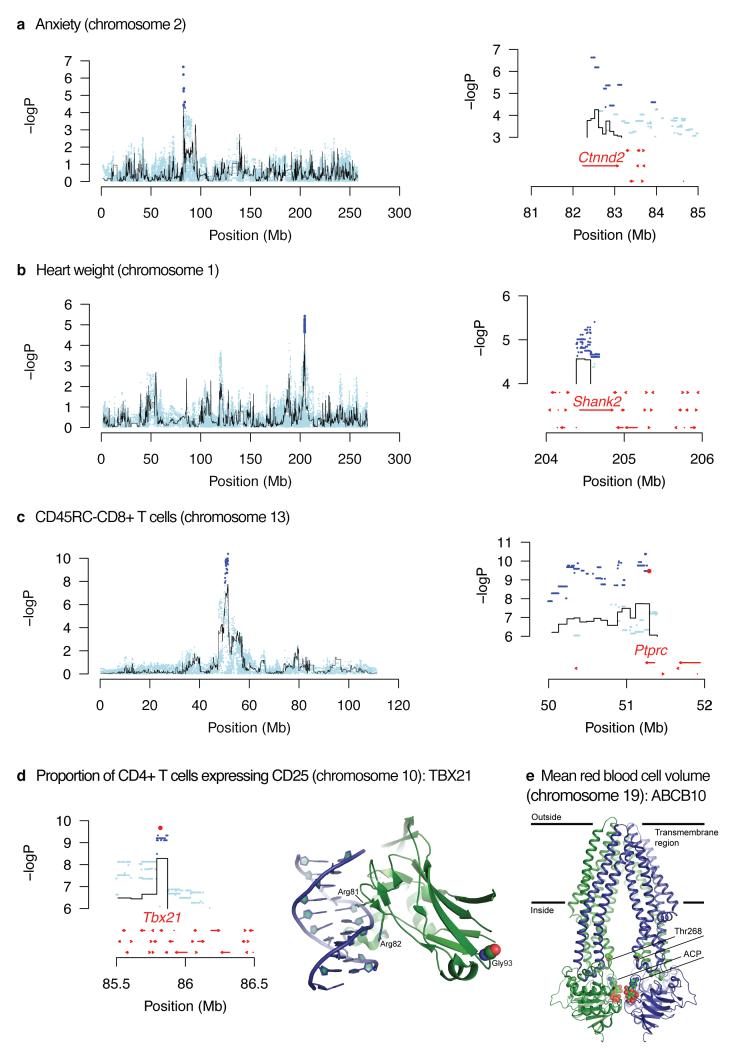
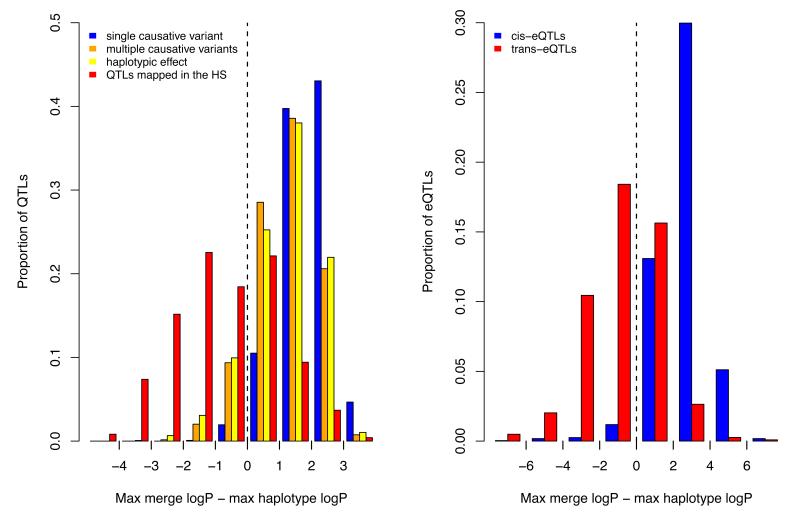
References
-
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–31. - PubMed
-
- Hansen C, Spuhler K. Development of the National Institues of Health genetically heterogeneous rat stock. Alcoholism, Clinical and Experimental Research. 1984;8:477–479. - PubMed
-
- Talbot CJ, et al. High-resolution mapping of quantitative trait loci in outbred mice. Nature Genetics. 1999;21:305–308. - PubMed
Publication types
MeSH terms
Grants and funding
- MC_U120061454/MRC_/Medical Research Council/United Kingdom
- 089269/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- RG/07/005/23633/BHF_/British Heart Foundation/United Kingdom
- BHFRG/07/005/23633/BHF_/British Heart Foundation/United Kingdom
- A10976/CRUK_/Cancer Research UK/United Kingdom
- U54 HG003273/HG/NHGRI NIH HHS/United States
- G0900084/MRC_/Medical Research Council/United Kingdom
- G9900061/MRC_/Medical Research Council/United Kingdom
- 083573/Z/07/Z/WT_/Wellcome Trust/United Kingdom
- R01 AR047822/AR/NIAMS NIH HHS/United States
- 083573/WT_/Wellcome Trust/United Kingdom
- 090532/WT_/Wellcome Trust/United Kingdom
- 13031/CRUK_/Cancer Research UK/United Kingdom
- 089269/WT_/Wellcome Trust/United Kingdom
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
