

Efficient moment-based inference of admixture parameters and sources of gene flow
- PMID: 23709261
- PMCID: PMC3708505
- DOI: 10.1093/molbev/mst099
Efficient moment-based inference of admixture parameters and sources of gene flow
Abstract
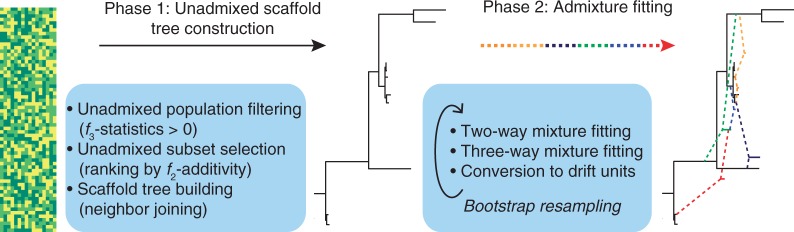
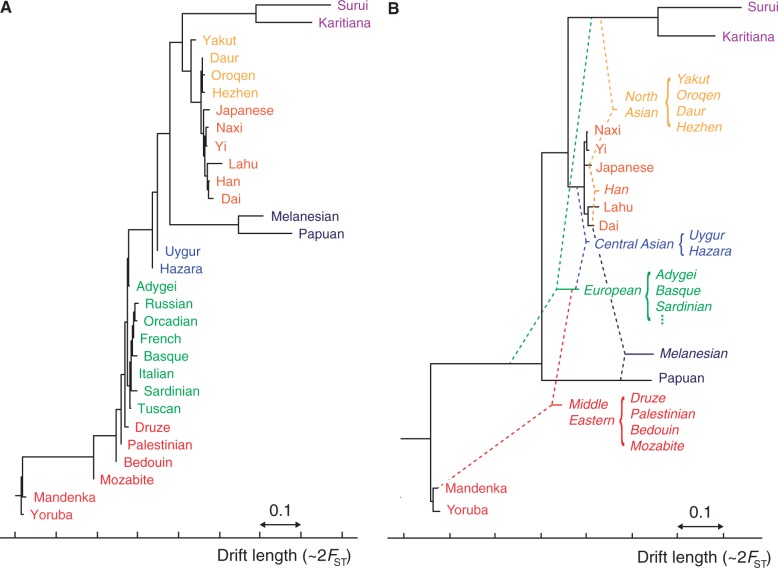
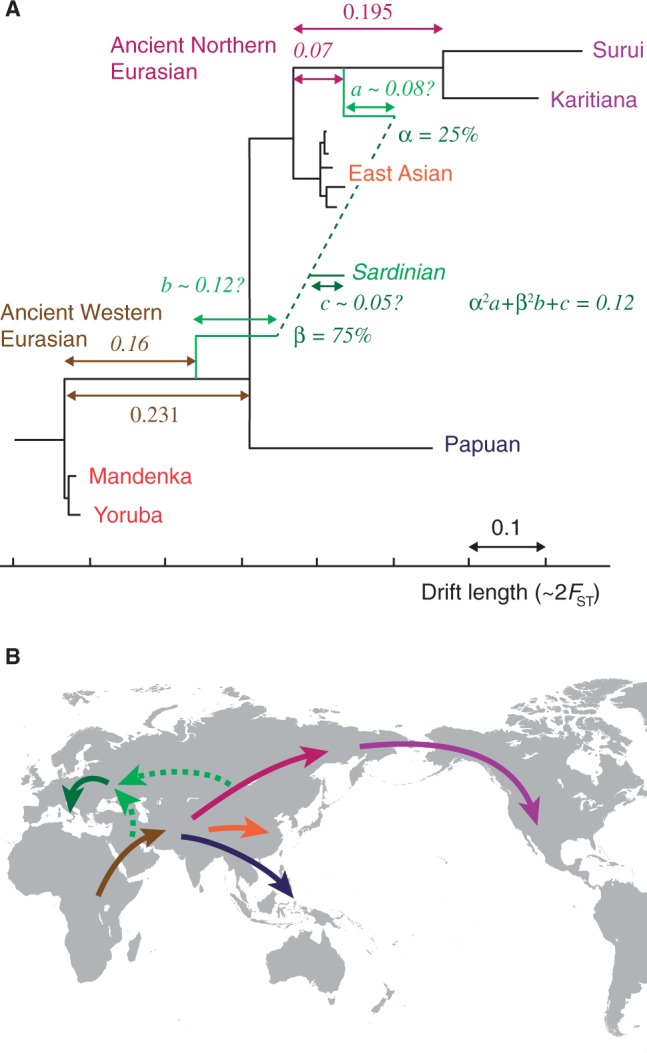
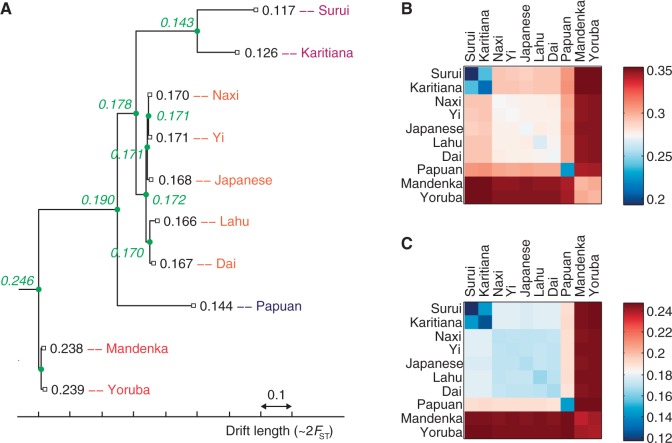
The recent explosion in available genetic data has led to significant advances in understanding the demographic histories of and relationships among human populations. It is still a challenge, however, to infer reliable parameter values for complicated models involving many populations. Here, we present MixMapper, an efficient, interactive method for constructing phylogenetic trees including admixture events using single nucleotide polymorphism (SNP) genotype data. MixMapper implements a novel two-phase approach to admixture inference using moment statistics, first building an unadmixed scaffold tree and then adding admixed populations by solving systems of equations that express allele frequency divergences in terms of mixture parameters. Importantly, all features of the model, including topology, sources of gene flow, branch lengths, and mixture proportions, are optimized automatically from the data and include estimates of statistical uncertainty. MixMapper also uses a new method to express branch lengths in easily interpretable drift units. We apply MixMapper to recently published data for Human Genome Diversity Cell Line Panel individuals genotyped on a SNP array designed especially for use in population genetics studies, obtaining confident results for 30 populations, 20 of them admixed. Notably, we confirm a signal of ancient admixture in European populations-including previously undetected admixture in Sardinians and Basques-involving a proportion of 20-40% ancient northern Eurasian ancestry.
Keywords: admixture; genetic drift; human populations; moment statistics.
Figures
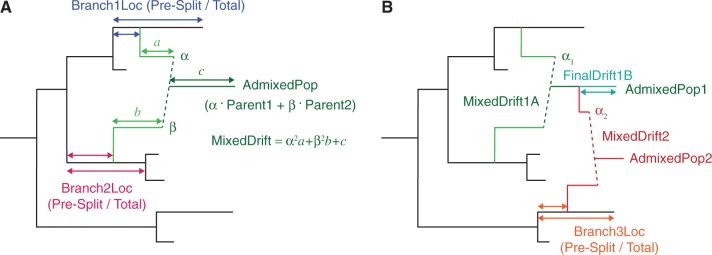
 is the proportion from Branch2); and MixedDrift is the linear combination of drift lengths
is the proportion from Branch2); and MixedDrift is the linear combination of drift lengths  . (B) A three-way mixture: here AdmixedPop2 is modeled as an admixture between AdmixedPop1 and Branch3. There are now four additional parameters; three are analogous to the above, namely, Branch3Loc,
. (B) A three-way mixture: here AdmixedPop2 is modeled as an admixture between AdmixedPop1 and Branch3. There are now four additional parameters; three are analogous to the above, namely, Branch3Loc,  , and MixedDrift2. The remaining degree of freedom is the position of the split along the AdmixedPop1 branch, which divides MixedDrift into MixedDrift1A and FinalDrift1B.
, and MixedDrift2. The remaining degree of freedom is the position of the split along the AdmixedPop1 branch, which divides MixedDrift into MixedDrift1A and FinalDrift1B.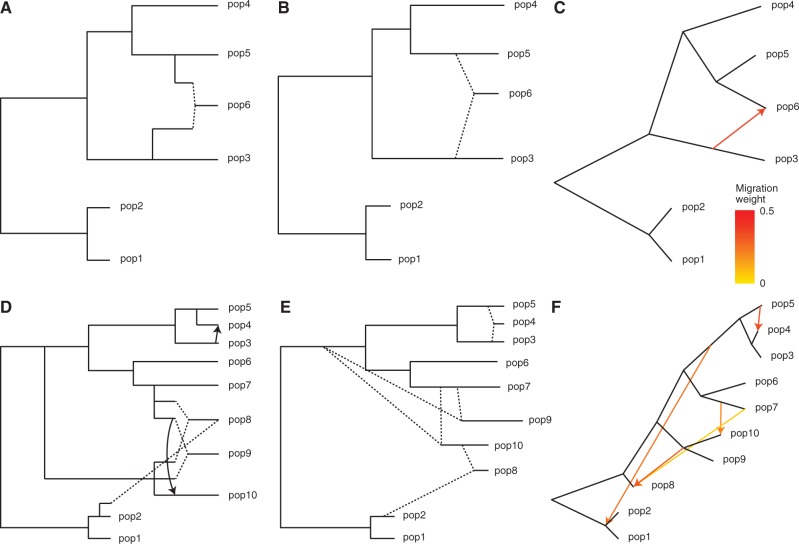
References
-
- Bramanti B, Thomas M, Haak W, et al. (11 co-authors) Genetic discontinuity between local hunter-gatherers and Central Europe’s first farmers. Science. 2009;326:137–140. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
