

Harnessing the platelet signaling network to produce an optimal hemostatic response
- PMID: 23714305
- PMCID: PMC3787971
- DOI: 10.1016/j.hoc.2013.02.002
Harnessing the platelet signaling network to produce an optimal hemostatic response
Abstract
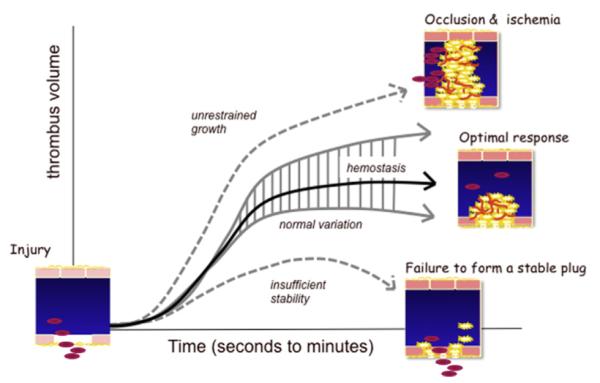
Once released into the circulation by megakaryocytes, circulating platelets can undergo rapid activation at sites of vascular injury and resist unwarranted activation, which can lead to heart attacks and strokes. Historically, the signaling mechanisms underlying the regulation of platelet activation have been approached as a collection of individual pathways unique to agonist. This review takes a different approach, casting platelet activation as the product of a signaling network, in which activating and restraining mechanisms interact in a flexible network that regulates platelet adhesiveness, cohesion between platelets, granule secretion, and the formation of a stable hemostatic thrombus.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
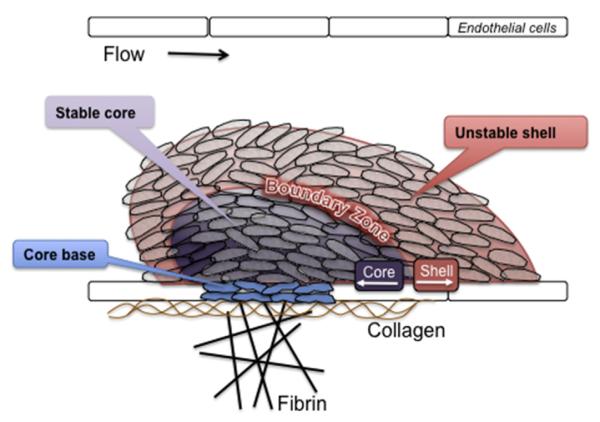
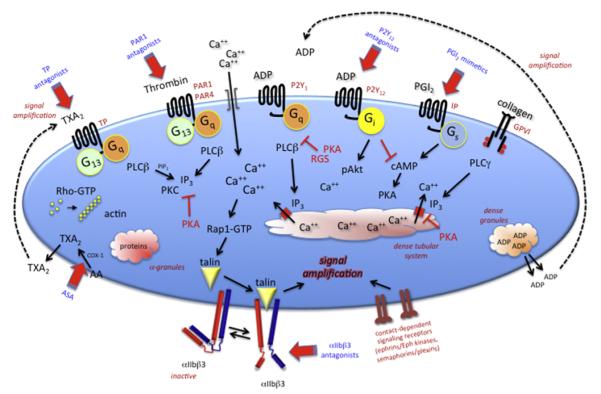
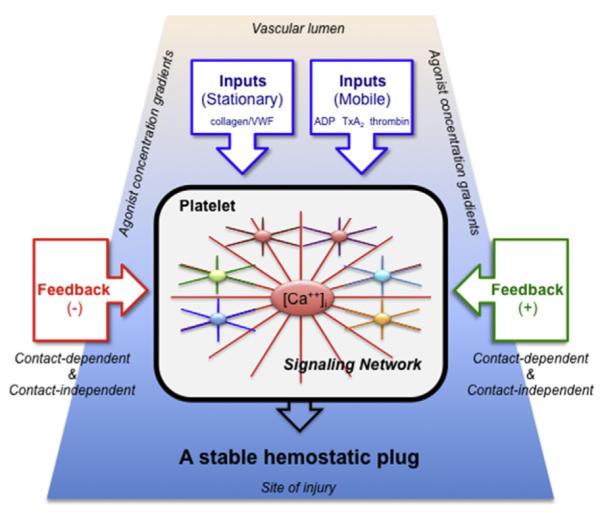
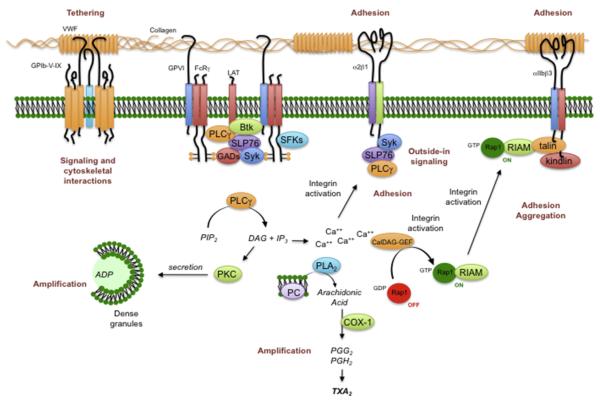
References
-
- Munnix IC, Cosemans JM, Auger JM, et al. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102:1149–56. - PubMed
-
- Nesbitt WS, Westein E, Tovar-Lopez FJ, et al. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–73. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
