

Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6
- PMID: 23714752
- PMCID: PMC3895628
- DOI: 10.1038/ejhg.2013.108
Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6
Abstract
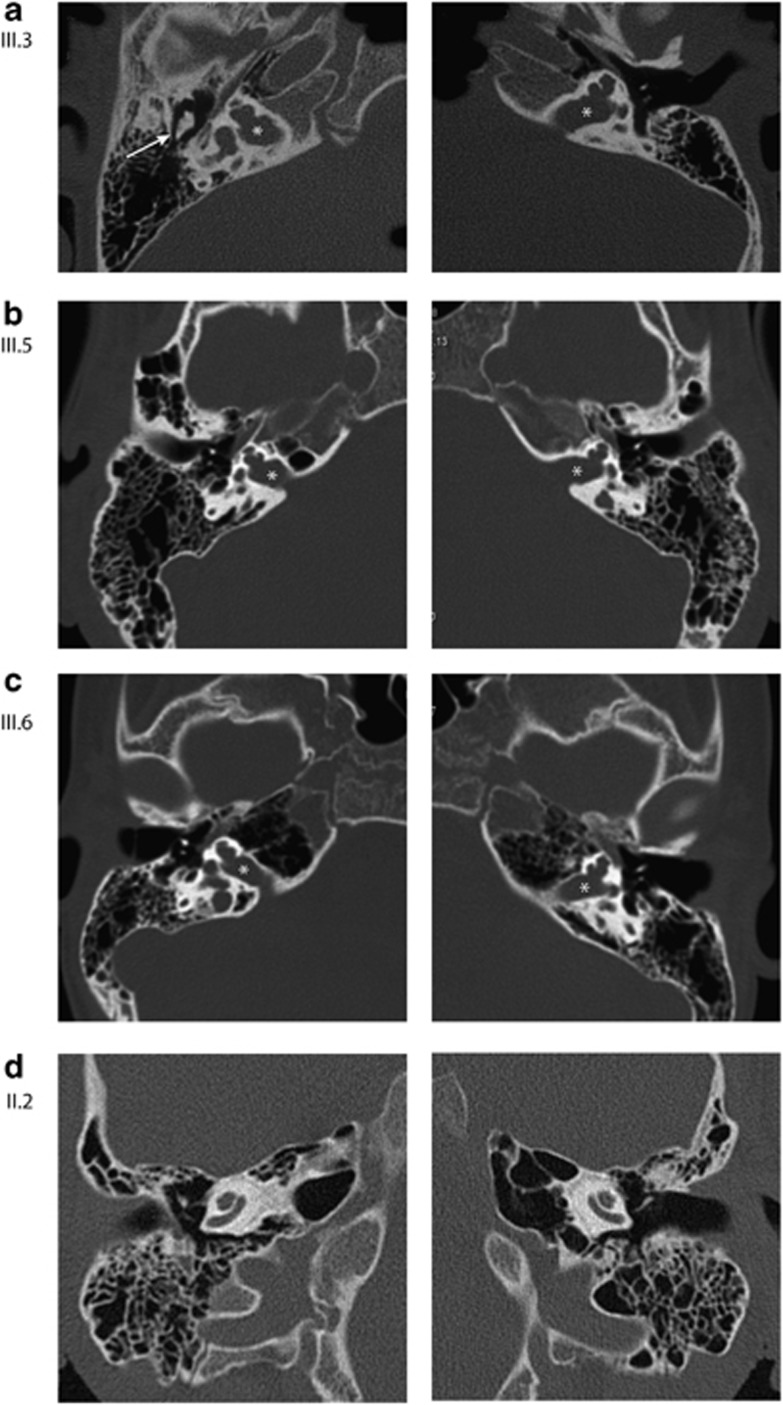
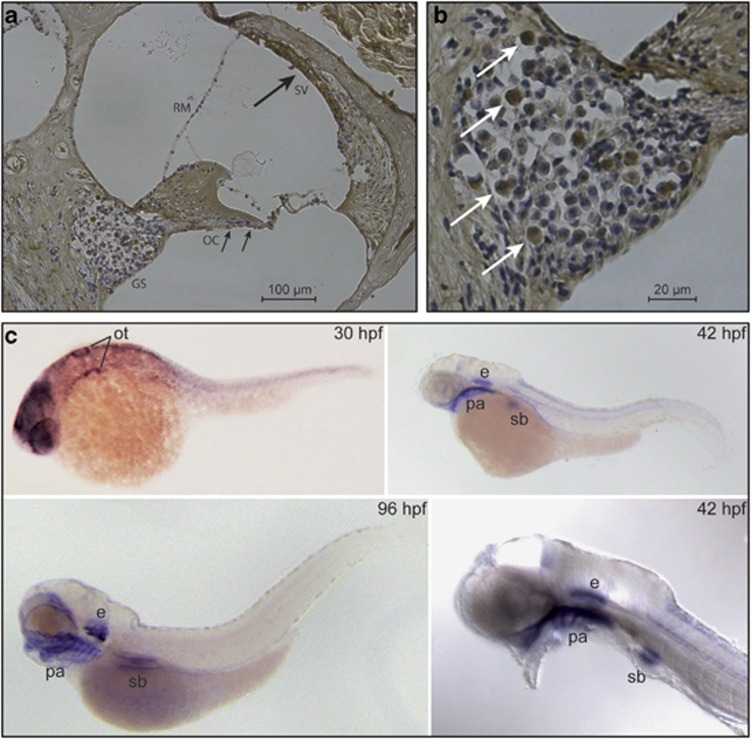
Hereditary hearing loss is the most common human sensorineural disorder. Genetic causes are highly heterogeneous, with mutations detected in >40 genes associated with nonsyndromic hearing loss, to date. Whereas autosomal recessive and autosomal dominant inheritance is prevalent, X-linked forms of nonsyndromic hearing impairment are extremely rare. Here, we present a Hungarian three-generation family with X-linked nonsyndromic congenital hearing loss and the underlying genetic defect. Next-generation sequencing and subsequent segregation analysis detected a missense mutation (c.1771G>A, p.Gly591Ser) in the type IV collagen gene COL4A6 in all affected family members. Bioinformatic analysis and expression studies support this substitution as being causative. COL4A6 encodes the alpha-6 chain of type IV collagen of basal membranes, which forms a heterotrimer with two alpha-5 chains encoded by COL4A5. Whereas mutations in COL4A5 and contiguous X-chromosomal deletions involving COL4A5 and COL4A6 are associated with X-linked Alport syndrome, a nephropathy associated with deafness and cataract, mutations in COL4A6 alone have not been related to any hereditary disease so far. Moreover, our index patient and other affected family members show normal renal and ocular function, which is not consistent with Alport syndrome, but with a nonsyndromic type of hearing loss. In situ hybridization and immunostaining demonstrated expression of the COL4A6 homologs in the otic vesicle of the zebrafish and in the murine inner ear, supporting its role in normal ear development and function. In conclusion, our results suggest COL4A6 as being the fourth gene associated with X-linked nonsyndromic hearing loss.
Figures


References
-
- Bayazit YA, Yilmaz M. An overview of hereditary hearing loss. ORL J Otorhinolaryngol Relat Spec. 2006;68:57–63. - PubMed
-
- Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clin Genet. 2007;71:379–391. - PubMed
-
- Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. - PubMed
-
- Tekin M, Arnos KS, Pandya A. Advances in hereditary deafness. Lancet. 2001;358:1082–1090. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
