

Computational analysis of bacterial RNA-Seq data
- PMID: 23716638
- PMCID: PMC3737546
- DOI: 10.1093/nar/gkt444
Computational analysis of bacterial RNA-Seq data
Abstract
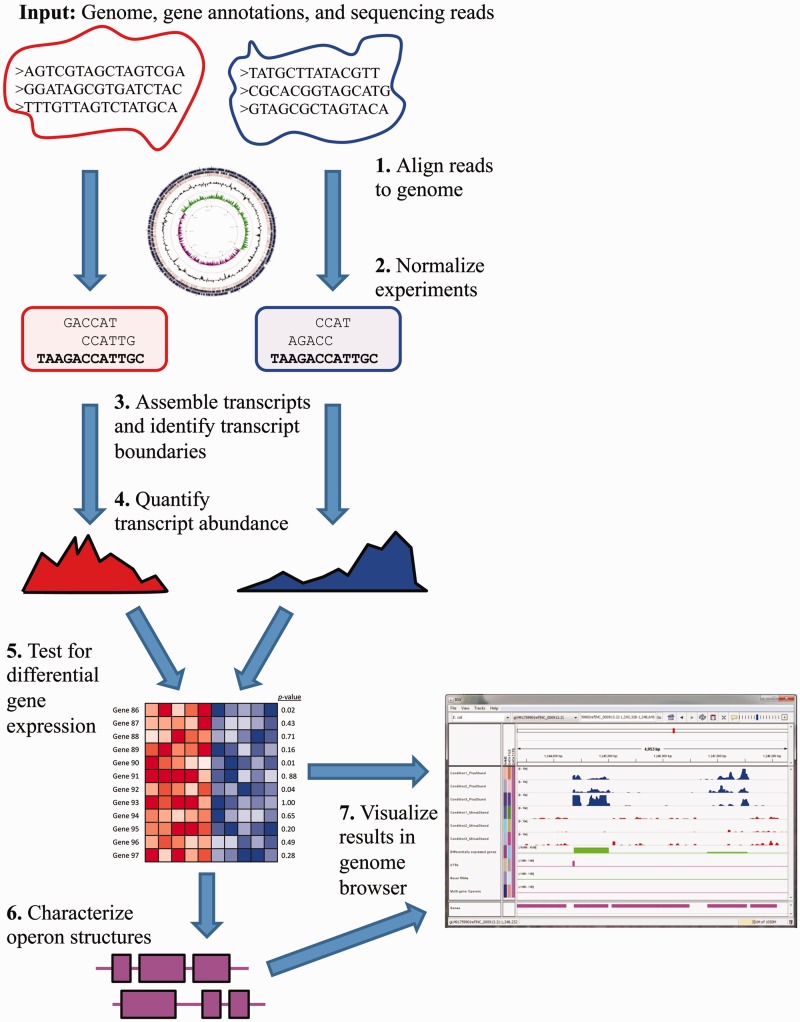
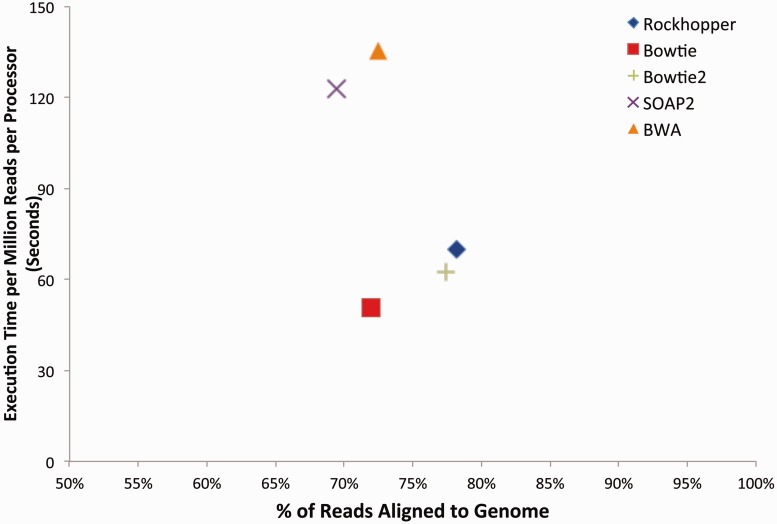
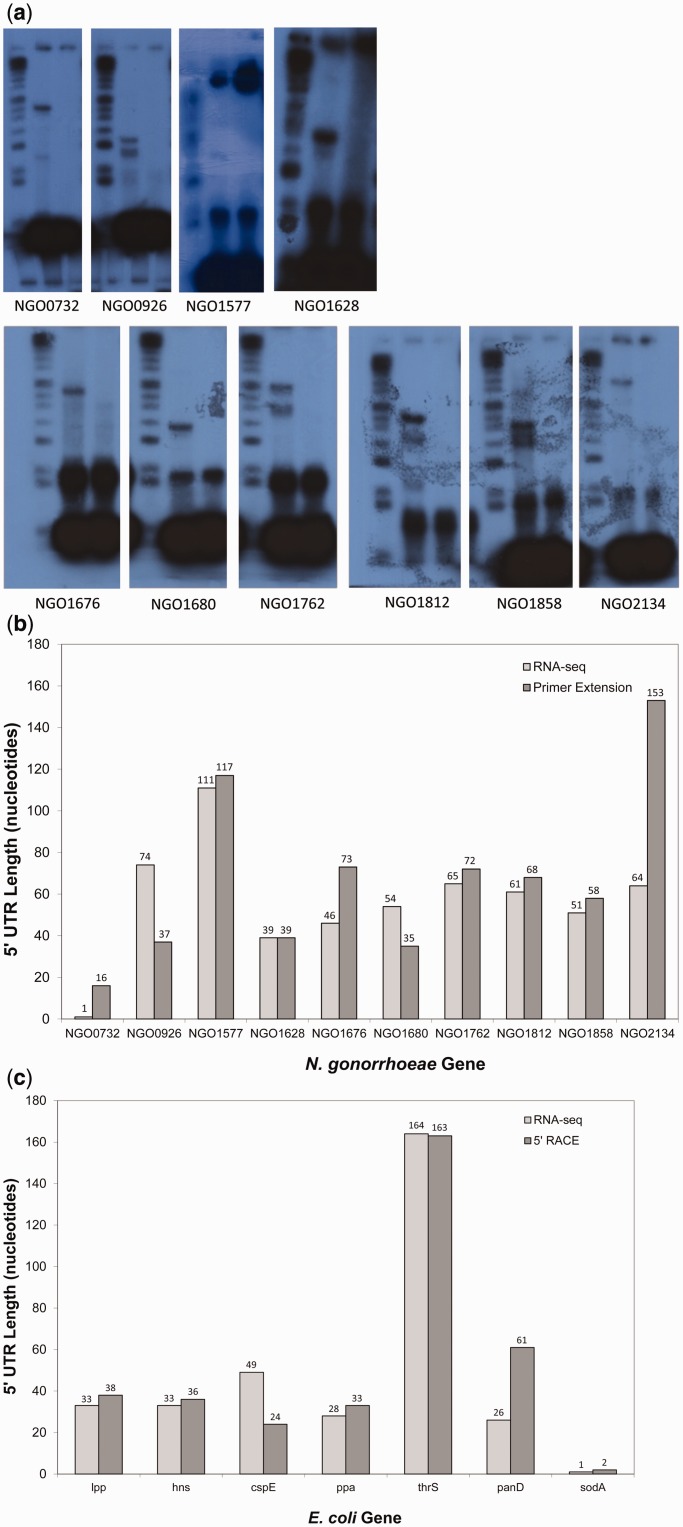
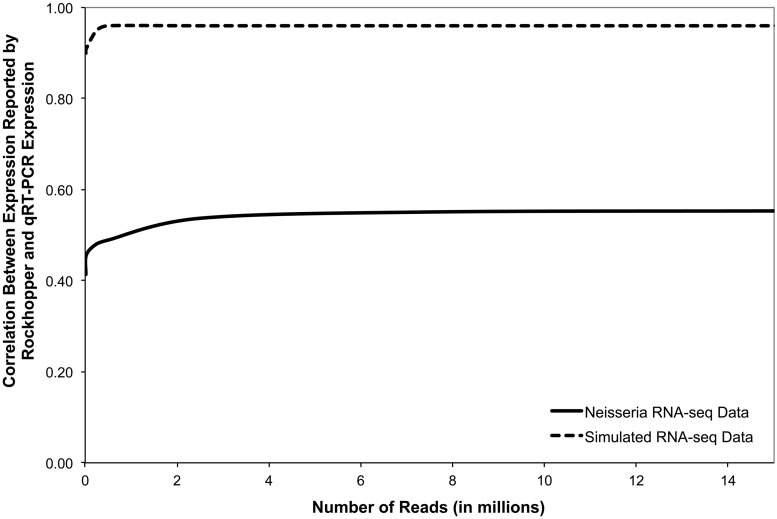
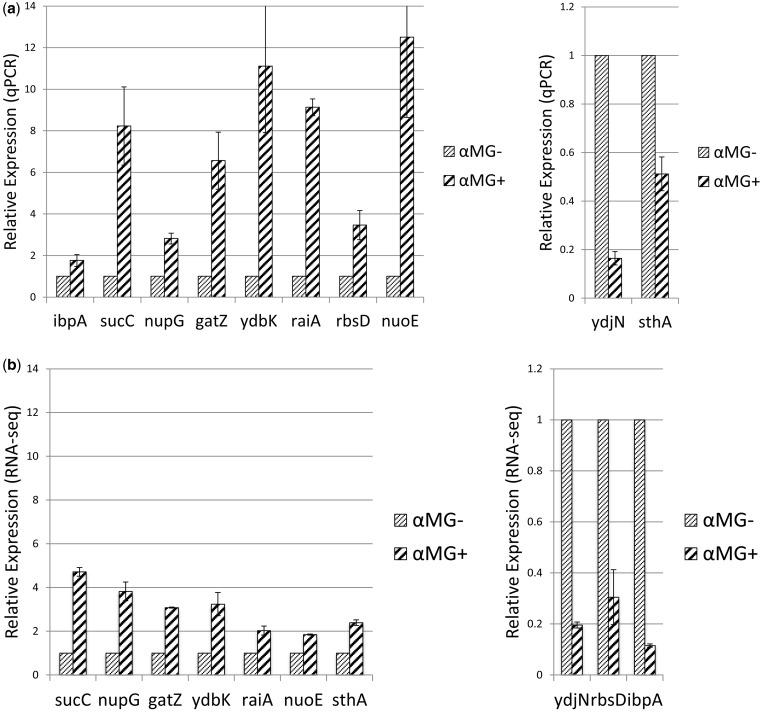
Recent advances in high-throughput RNA sequencing (RNA-seq) have enabled tremendous leaps forward in our understanding of bacterial transcriptomes. However, computational methods for analysis of bacterial transcriptome data have not kept pace with the large and growing data sets generated by RNA-seq technology. Here, we present new algorithms, specific to bacterial gene structures and transcriptomes, for analysis of RNA-seq data. The algorithms are implemented in an open source software system called Rockhopper that supports various stages of bacterial RNA-seq data analysis, including aligning sequencing reads to a genome, constructing transcriptome maps, quantifying transcript abundance, testing for differential gene expression, determining operon structures and visualizing results. We demonstrate the performance of Rockhopper using 2.1 billion sequenced reads from 75 RNA-seq experiments conducted with Escherichia coli, Neisseria gonorrhoeae, Salmonella enterica, Streptococcus pyogenes and Xenorhabdus nematophila. We find that the transcriptome maps generated by our algorithms are highly accurate when compared with focused experimental data from E. coli and N. gonorrhoeae, and we validate our system's ability to identify novel small RNAs, operons and transcription start sites. Our results suggest that Rockhopper can be used for efficient and accurate analysis of bacterial RNA-seq data, and that it can aid with elucidation of bacterial transcriptomes.
Figures


References
-
- Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010;11:9–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
