

Apoptosis and necrosis in the liver
- PMID: 23720337
- PMCID: PMC3867948
- DOI: 10.1002/cphy.c120020
Apoptosis and necrosis in the liver
Abstract
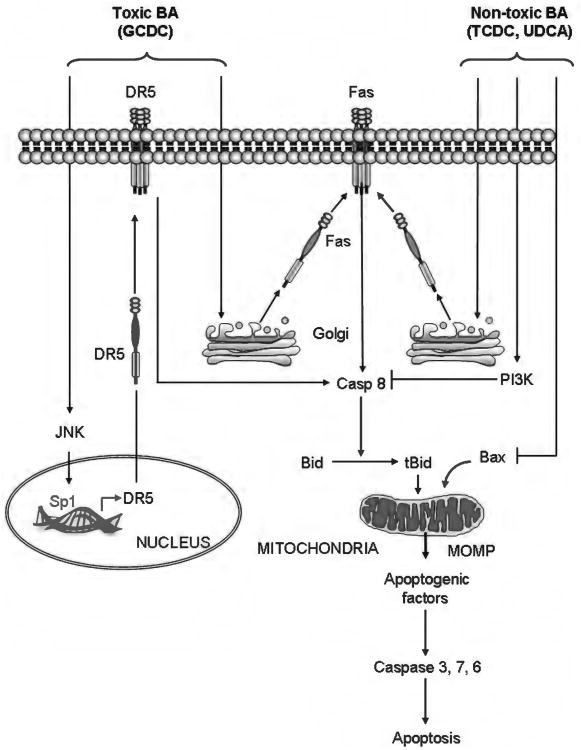
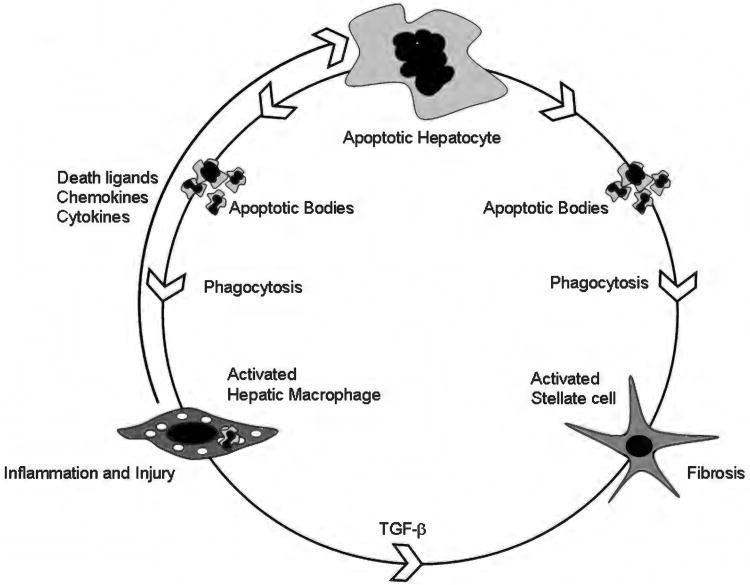
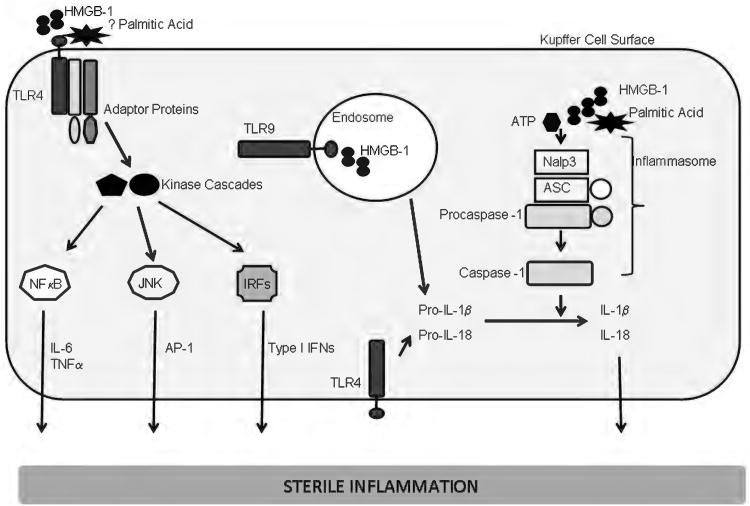
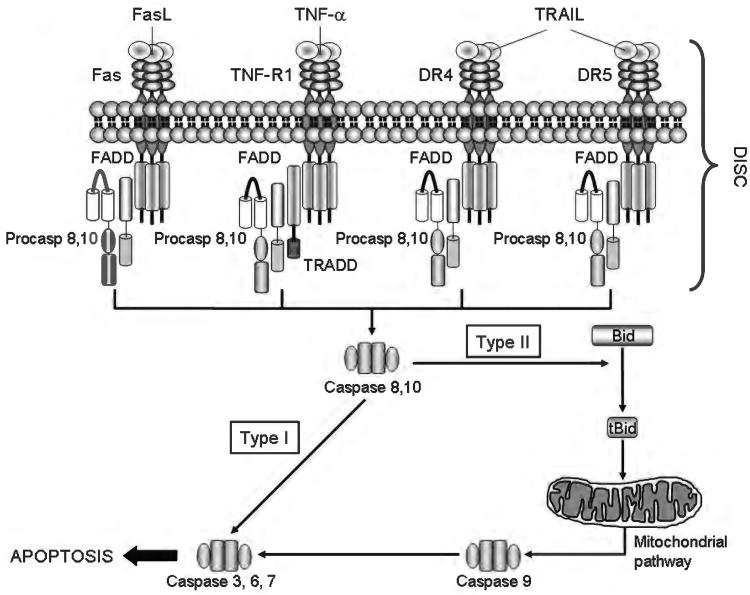
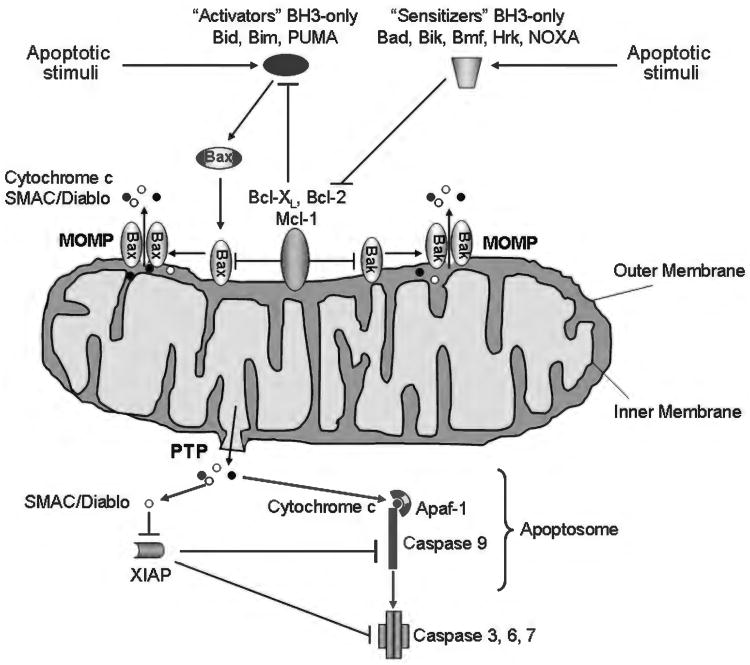
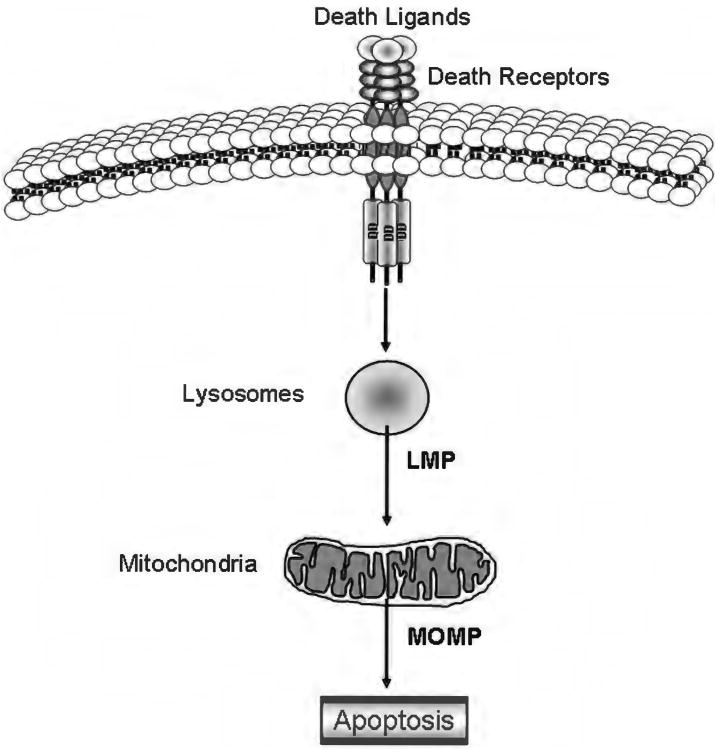
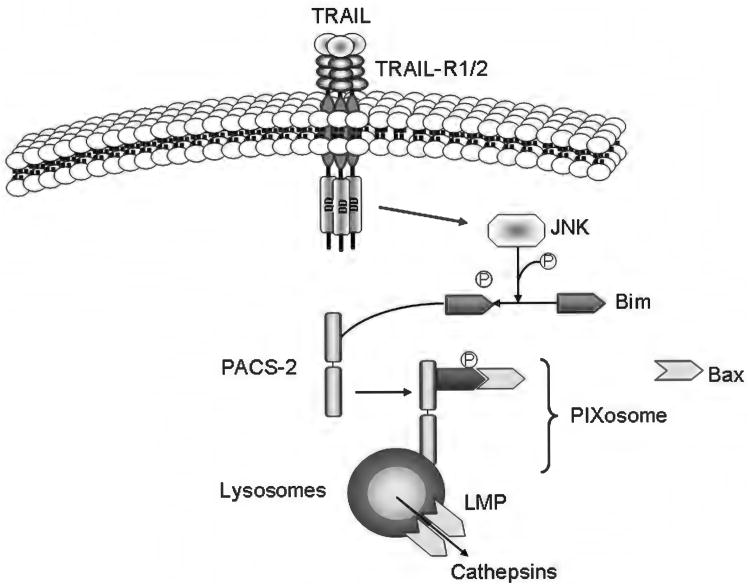
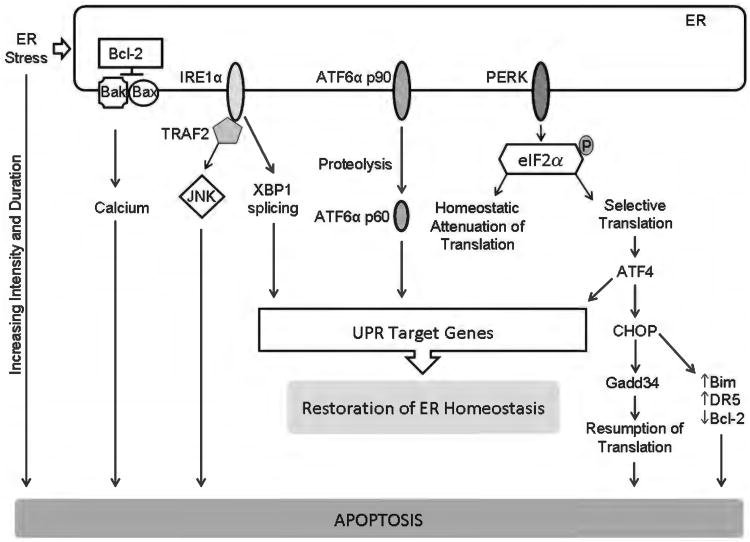
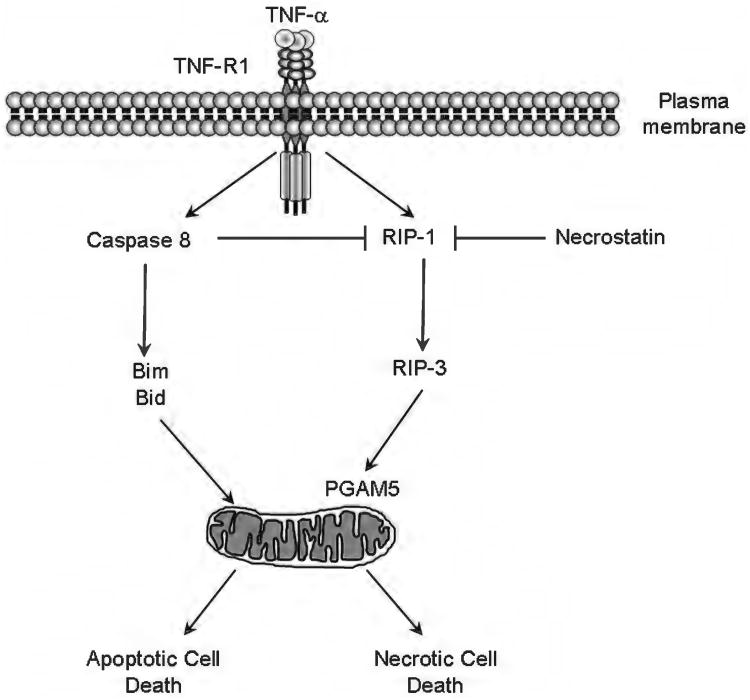
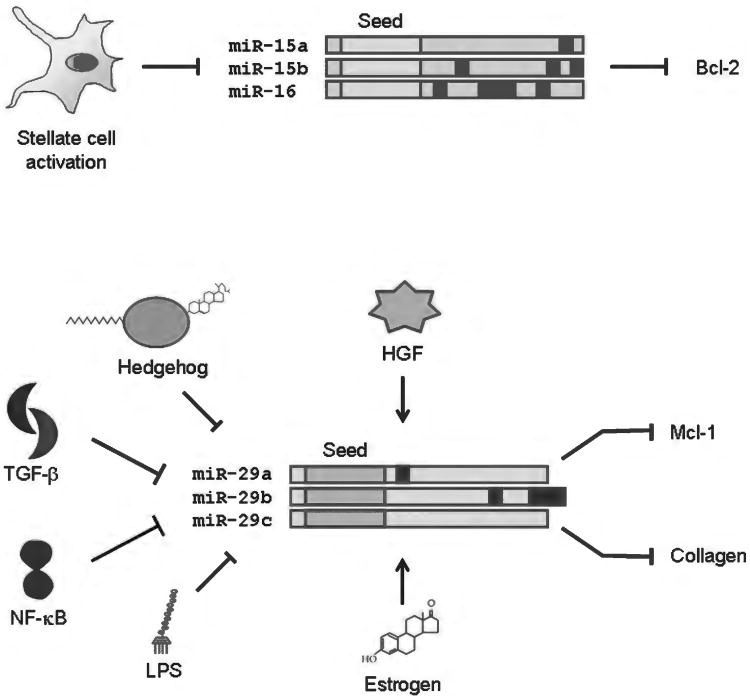
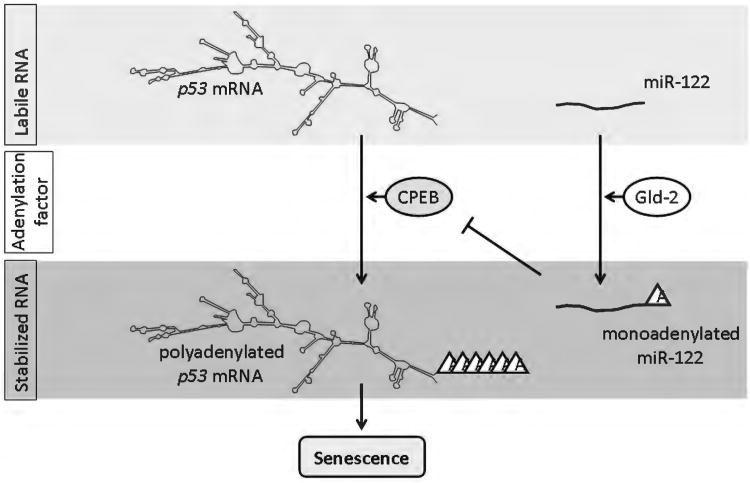
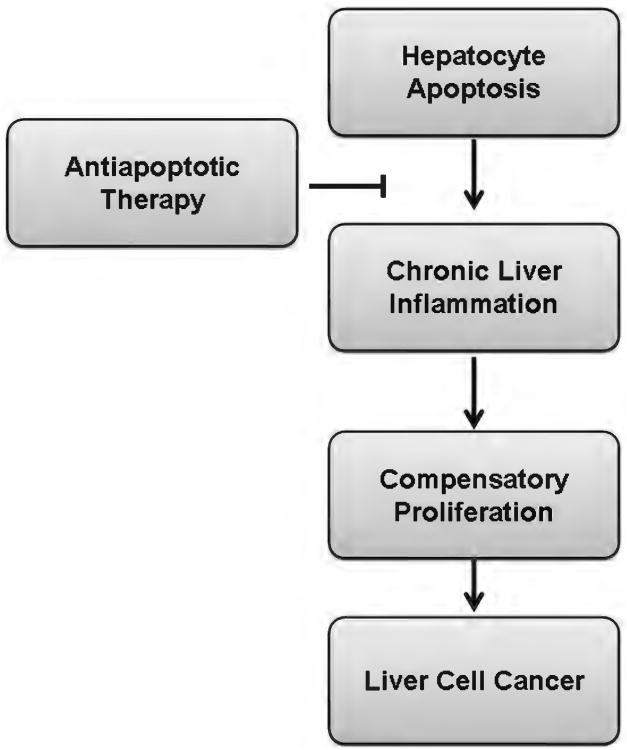
Because of its unique function and anatomical location, the liver is exposed to a multitude of toxins and xenobiotics, including medications and alcohol, as well as to infection by hepatotropic viruses, and therefore, is highly susceptible to tissue injury. Cell death in the liver occurs mainly by apoptosis or necrosis, with apoptosis also being the physiologic route to eliminate damaged or infected cells and to maintain tissue homeostasis. Liver cells, especially hepatocytes and cholangiocytes, are particularly susceptible to death receptor-mediated apoptosis, given the ubiquitous expression of the death receptors in the organ. In a quite unique way, death receptor-induced apoptosis in these cells is mediated by both mitochondrial and lysosomal permeabilization. Signaling between the endoplasmic reticulum and the mitochondria promotes hepatocyte apoptosis in response to excessive free fatty acid generation during the metabolic syndrome. These cell death pathways are partially regulated by microRNAs. Necrosis in the liver is generally associated with acute injury (i.e., ischemia/reperfusion injury) and has been long considered an unregulated process. Recently, a new form of "programmed" necrosis (named necroptosis) has been described: the role of necroptosis in the liver has yet to be explored. However, the minimal expression of a key player in this process in the liver suggests this form of cell death may be uncommon in liver diseases. Because apoptosis is a key feature of so many diseases of the liver, therapeutic modulation of liver cell death holds promise. An updated overview of these concepts is given in this article.
Figures
References
-
- Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: Processes in inflammatory networks—a review. Liver Transplant. 2010;16:1016–1032. - PubMed
-
- Adachi M, Suematsu S, Kondo T, Ogasawara J, Tanaka T, Yoshida N, Nagata S. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat Genet. 1995;11:294–300. - PubMed
-
- Afford SC, Ahmed-Choudhury J, Randhawa S, Russell C, Youster J, Crosby HA, Eliopoulos A, Hubscher SG, Young LS, Adams DH. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J. 2001;15:2345–2354. - PubMed
-
- Akazawa Y, Gores GJ. Death receptor-mediated liver injury. Semin Liver Dis. 2007;27:327–338. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
