

Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells
- PMID: 23720344
- PMCID: PMC3742869
- DOI: 10.1152/ajprenal.00160.2013
Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells
Abstract
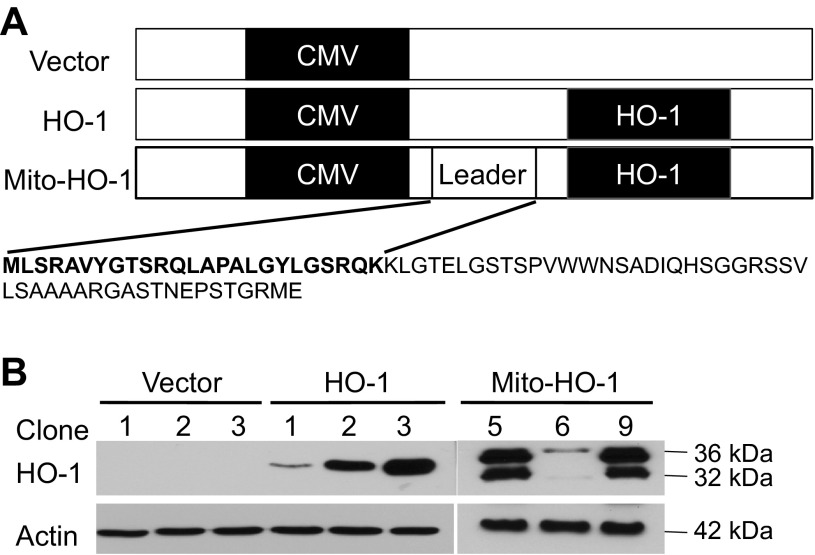
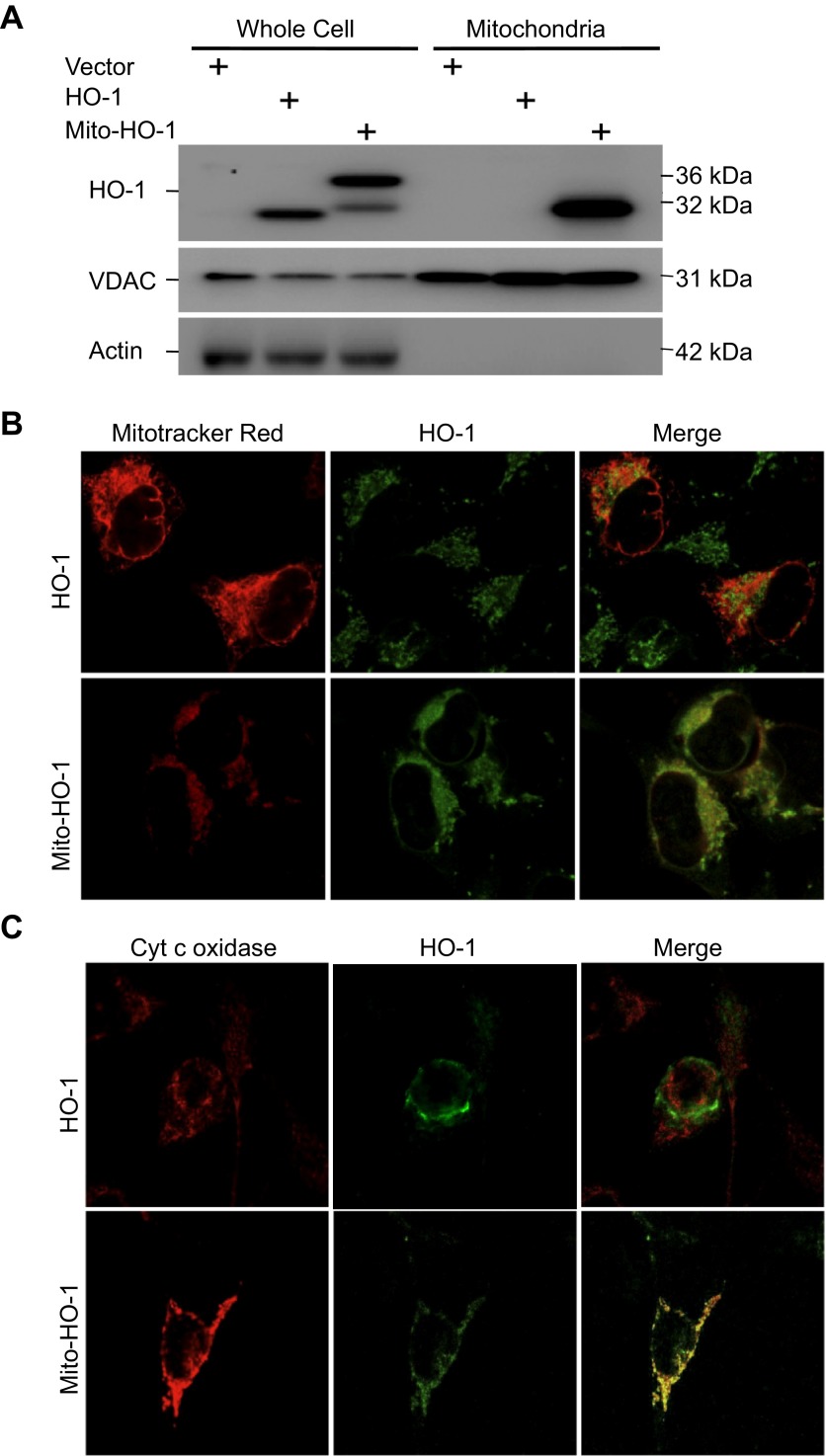
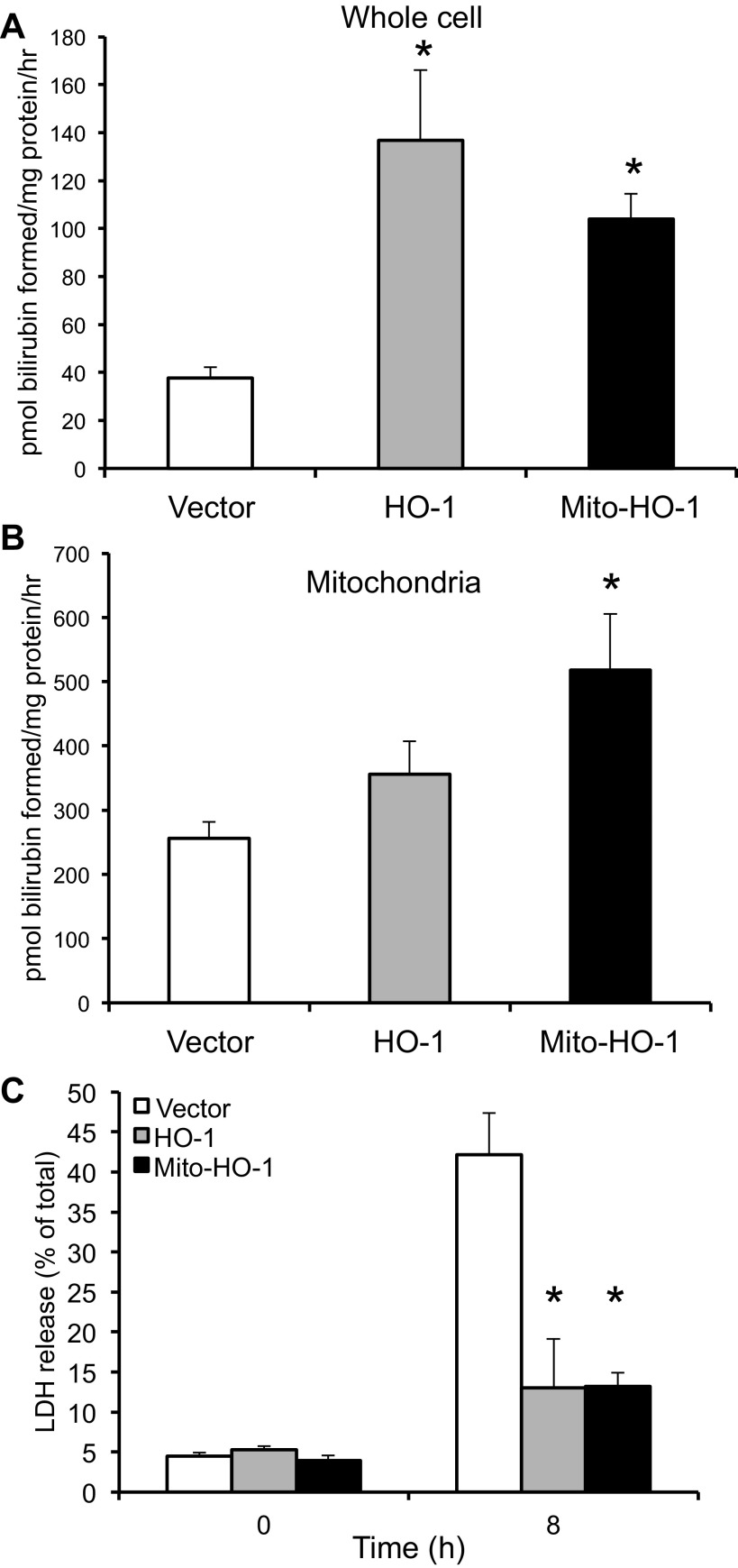
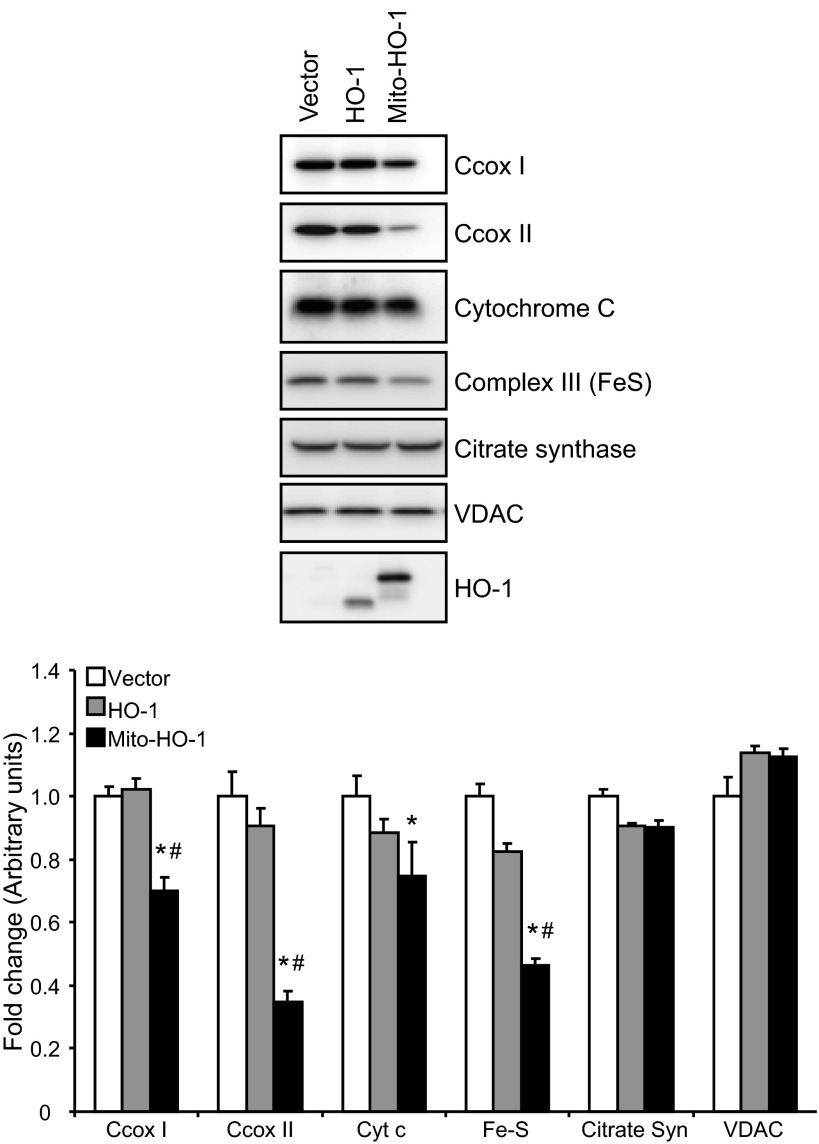
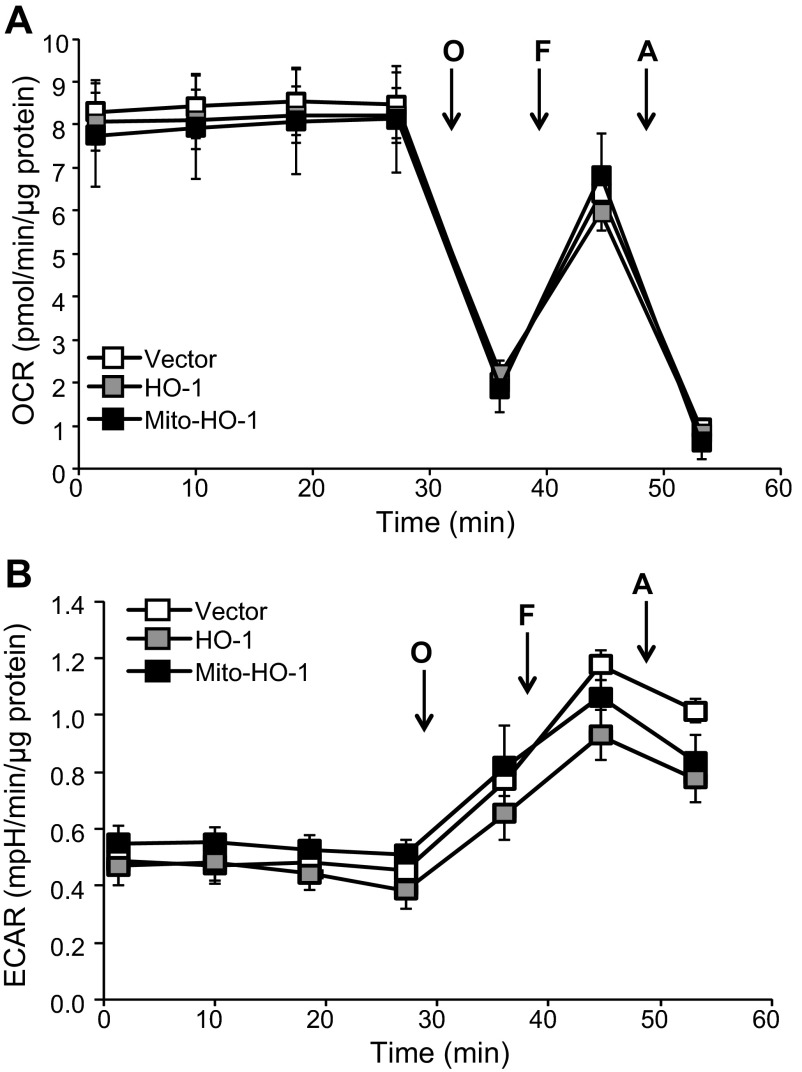
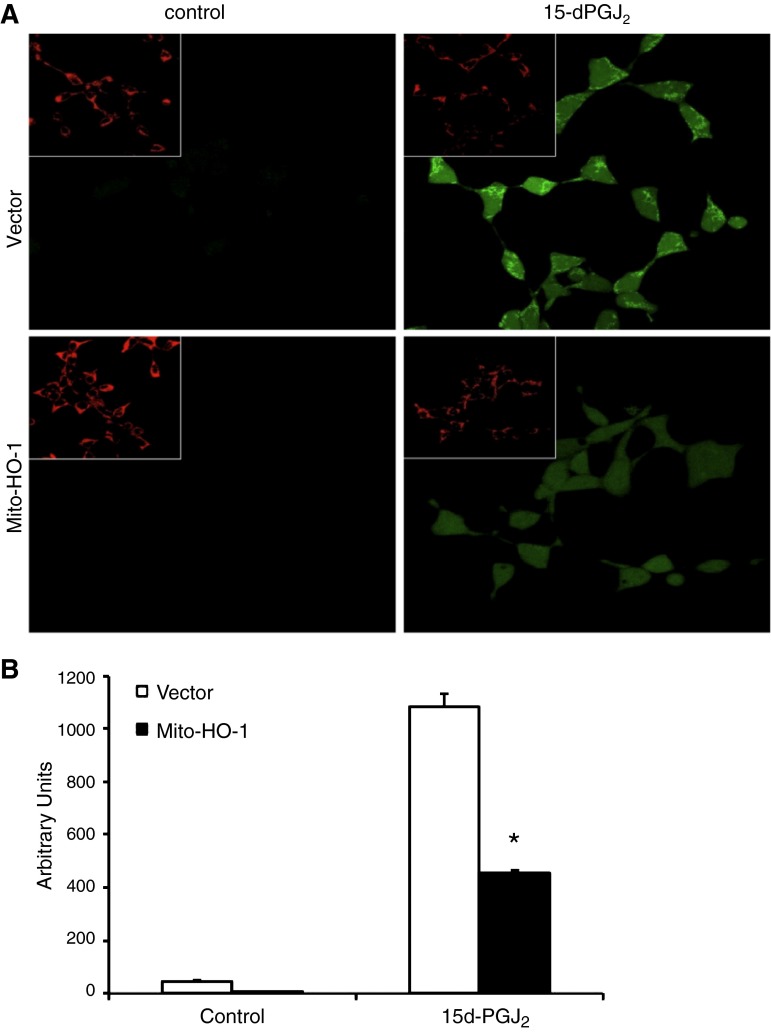
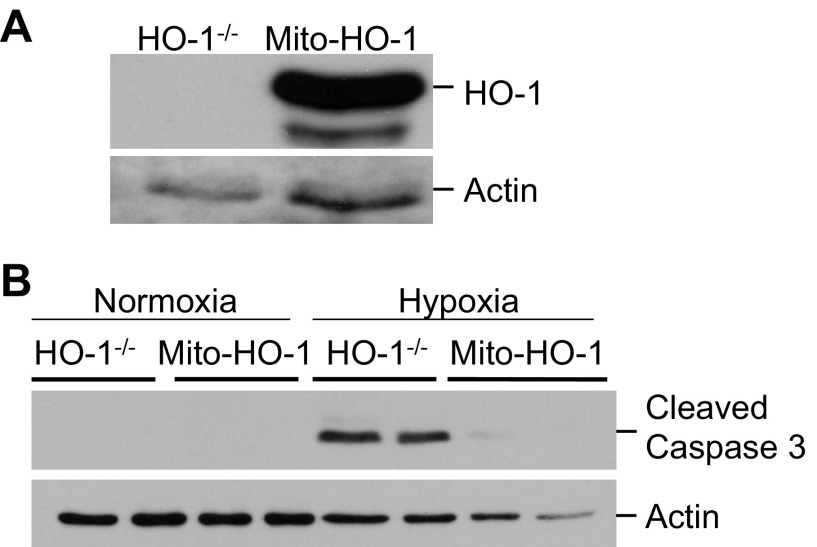
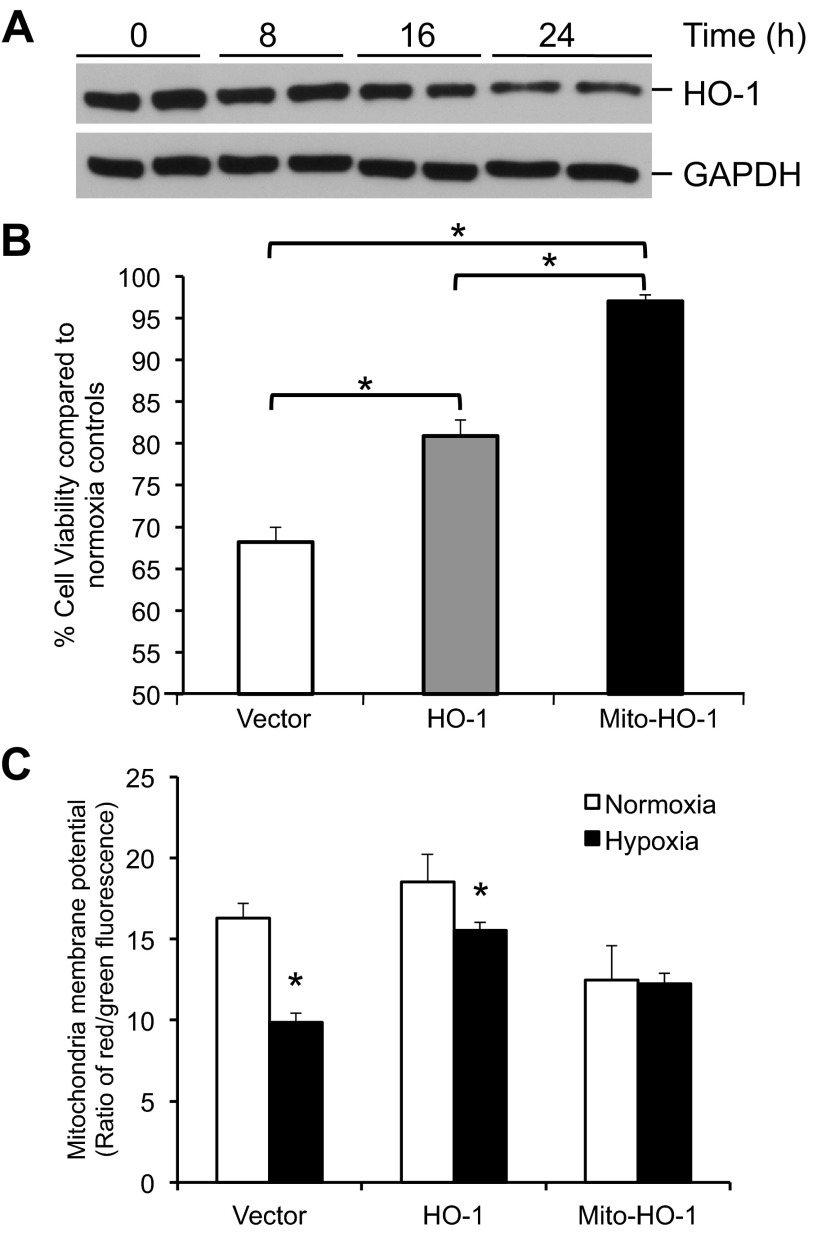
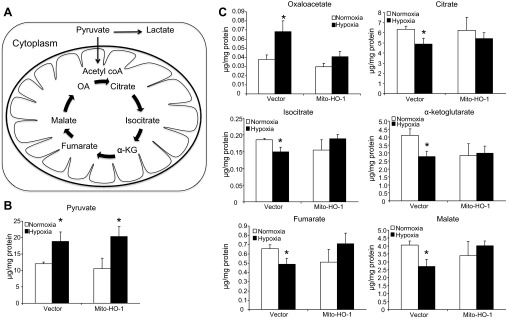
Mitochondria are both a source and target of the actions of reactive oxygen species and possess a complex system of inter-related antioxidants that control redox signaling and protect against oxidative stress. Interestingly, the antioxidant enzyme heme oxygenase-1 (HO-1) is not present in the mitochondria despite the fact that the organelle is the site of heme synthesis and contains multiple heme proteins. Detoxification of heme is an important protective mechanism since the reaction of heme with hydrogen peroxide generates pro-oxidant ferryl species capable of propagating oxidative stress and ultimately cell death. We therefore hypothesized that a mitochondrially localized HO-1 would be cytoprotective. To test this, we generated a mitochondria-targeted HO-1 cell line by transfecting HEK293 cells with a plasmid construct containing the manganese superoxide dismutase mitochondria leader sequence fused to HO-1 cDNA (Mito-HO-1). Nontargeted HO-1-overexpressing cells were generated by transfecting HO-1 cDNA (HO-1) or empty vector (Vector). Mitochondrial localization of HO-1 with increased HO activity in the mitochondrial fraction of Mito-HO-1 cells was observed, but a significant decrease in the expression of heme-containing proteins occurred in these cells. Both cytosolic HO-1- and Mito-HO-1-expressing cells were protected against hypoxia-dependent cell death and loss of mitochondrial membrane potential, but these effects were more pronounced with Mito-HO-1. Furthermore, decrement in production of tricarboxylic acid cycle intermediates following hypoxia was significantly mitigated in Mito-HO-1 cells. These data suggest that specific mitochondrially targeted HO-1 under acute pathological conditions may have beneficial effects, but the selective advantage of long-term expression is constrained by a negative impact on the synthesis of heme-containing mitochondrial proteins.
Keywords: heme; hypoxia.
Figures
References
-
- Baliga R, Ueda N, Walker PD, Shah SV. Oxidant mechanisms in toxic acute renal failure. Drug Metab Rev 31: 971–997, 1999 - PubMed
-
- Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med 32: 370–374, 2002 - PubMed
-
- Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med 33: 755–764, 2002 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases