

A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin
- PMID: 23720495
- PMCID: PMC3766181
- DOI: 10.1093/hmg/ddt242
A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin
Abstract
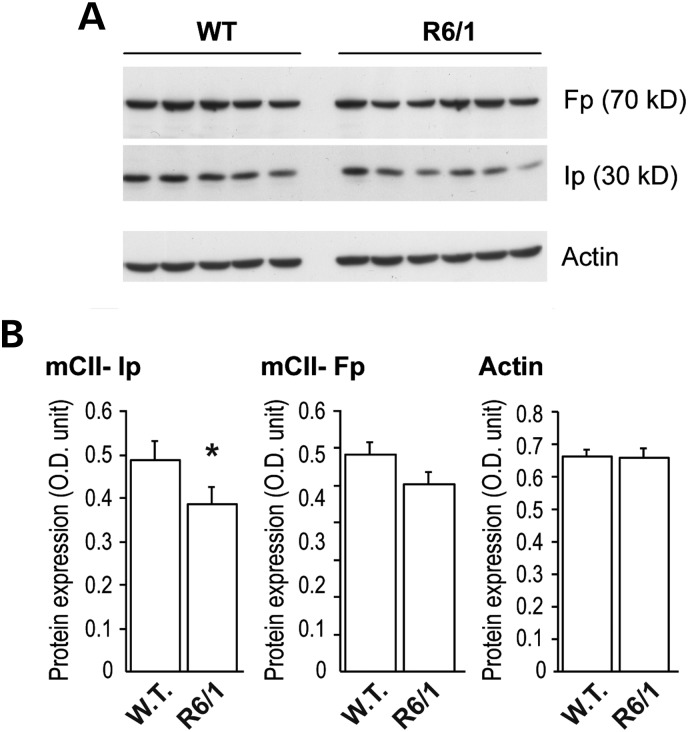
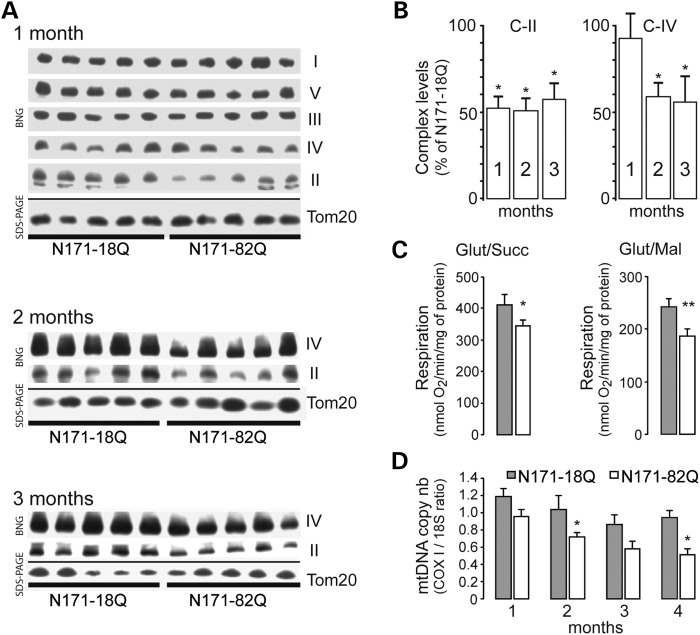
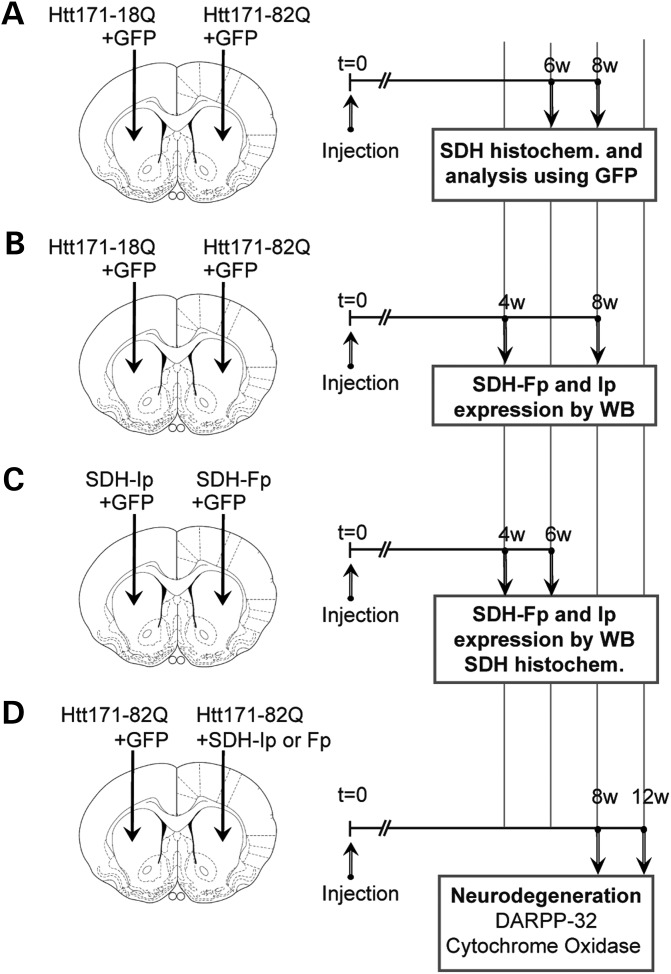
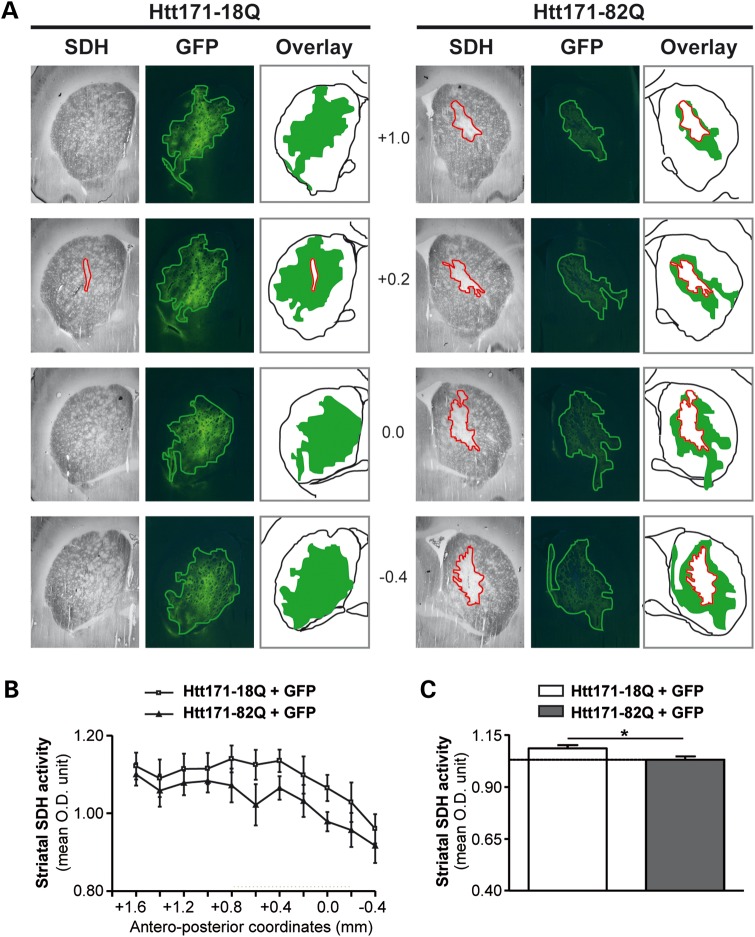
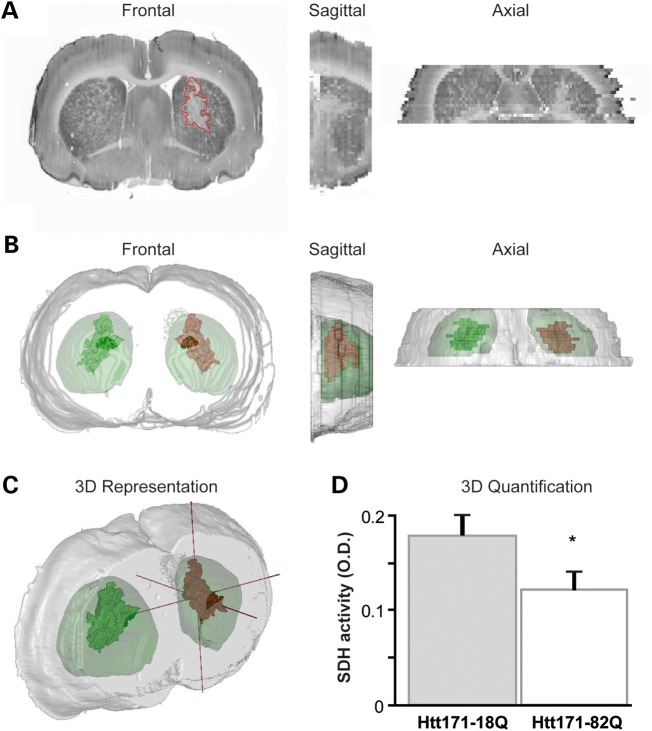
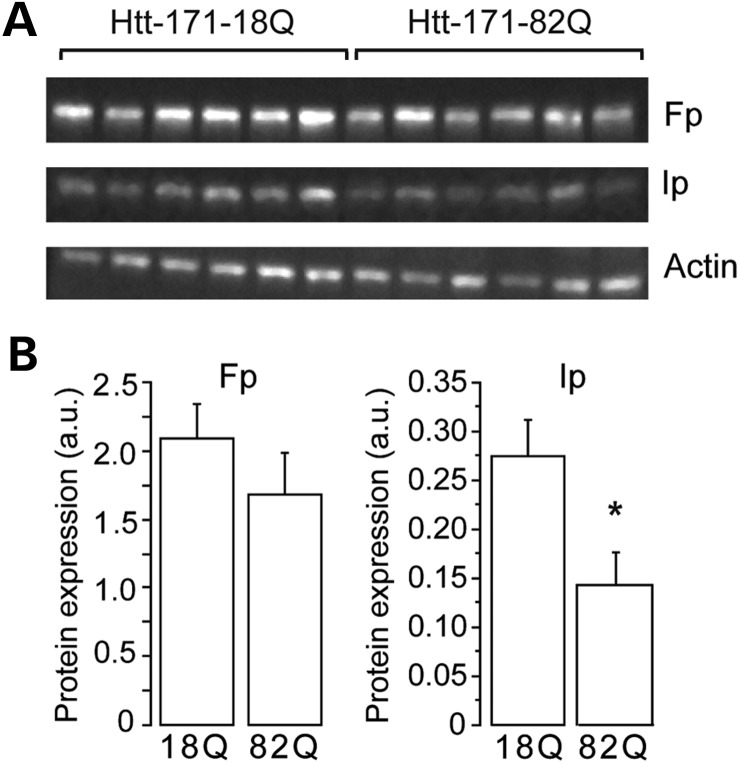
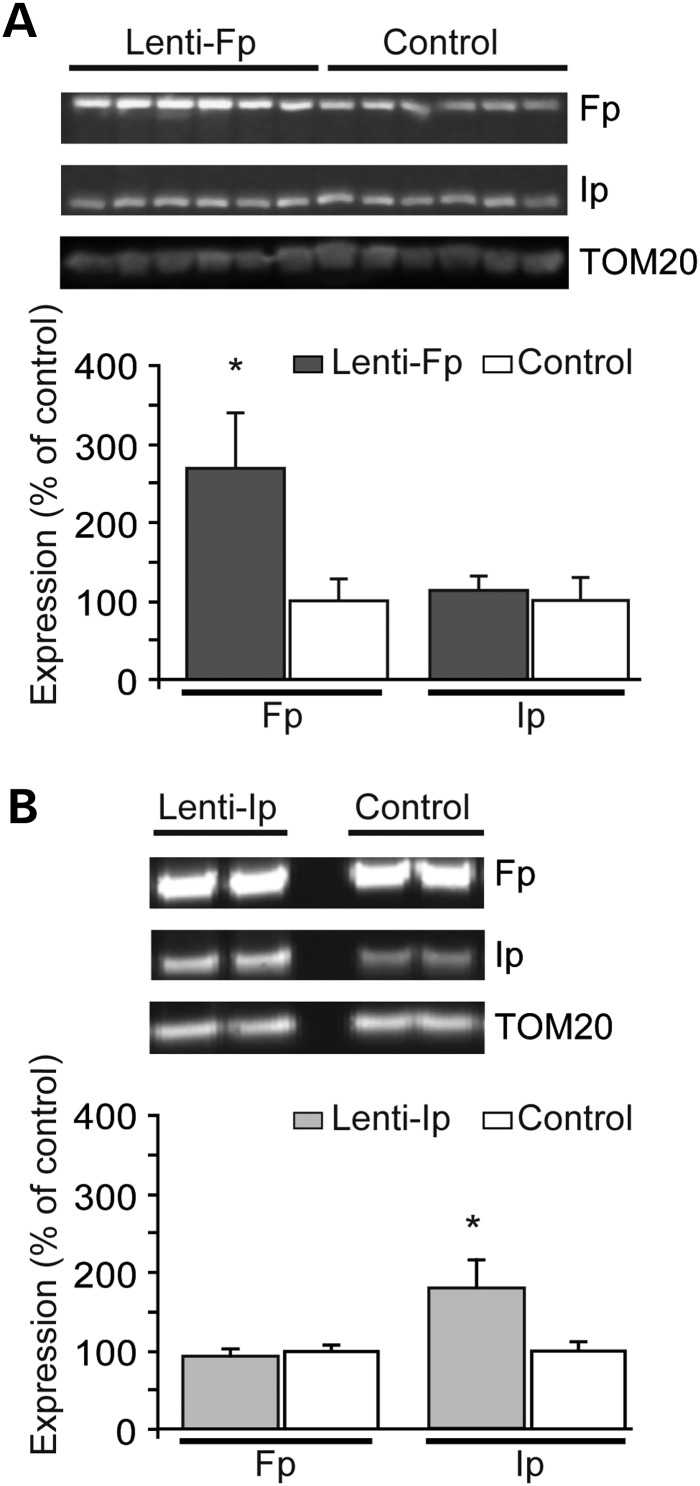
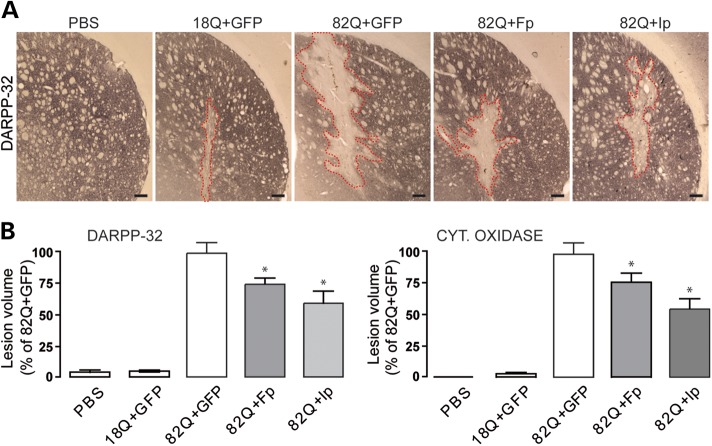
Huntington's disease (HD) is a neurodegenerative disorder caused by an abnormal expansion of a CAG repeat encoding a polyglutamine tract in the huntingtin (Htt) protein. The mutation leads to neuronal death through mechanisms which are still unknown. One hypothesis is that mitochondrial defects may play a key role. In support of this, the activity of mitochondrial complex II (C-II) is preferentially reduced in the striatum of HD patients. Here, we studied C-II expression in different genetic models of HD expressing N-terminal fragments of mutant Htt (mHtt). Western blot analysis showed that the expression of the 30 kDa Iron-Sulfur (Ip) subunit of C-II was significantly reduced in the striatum of the R6/1 transgenic mice, while the levels of the FAD containing catalytic 70 kDa subunit (Fp) were not significantly changed. Blue native gel analysis showed that the assembly of C-II in mitochondria was altered early in N171-82Q transgenic mice. Early loco-regional reduction in C-II activity and Ip protein expression was also demonstrated in a rat model of HD using intrastriatal injection of lentiviral vectors encoding mHtt. Infection of the rat striatum with a lentiviral vector coding the C-II Ip or Fp subunits induced a significant overexpression of these proteins that led to significant neuroprotection of striatal neurons against mHtt neurotoxicity. These results obtained in vivo support the hypothesis that structural and functional alterations of C-II induced by mHtt may play a critical role in the degeneration of striatal neurons in HD and that mitochondrial-targeted therapies may be useful in its treatment.
Figures
Comment in
-
Epigenetics of Huntington's disease.J Neurol. 2013 Nov;260(11):2938-41. doi: 10.1007/s00415-013-7158-x. J Neurol. 2013. PMID: 24141735 No abstract available.
References
-
- Damiano M., Galvan L., Deglon N., Brouillet E. Mitochondria in Huntington's disease. Biochim. Biophys. Acta. 2010;1802:52–61. - PubMed
-
- Harper P.S. Huntington's Disease. WB Saunders Company Ltd, London. 1991
-
- Myers R.H., Vonsattel J.P., Stevens T.J., Cupples L.A., Richardson E.P., Martin J.B., Bird E.D. Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology. 1988;38:341–347. - PubMed
-
- Sugars K.L., Rubinsztein D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
