

Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1α) negatively regulates β1 integrins
- PMID: 23720740
- PMCID: PMC3711292
- DOI: 10.1074/jbc.M113.455956
Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1α) negatively regulates β1 integrins
Abstract
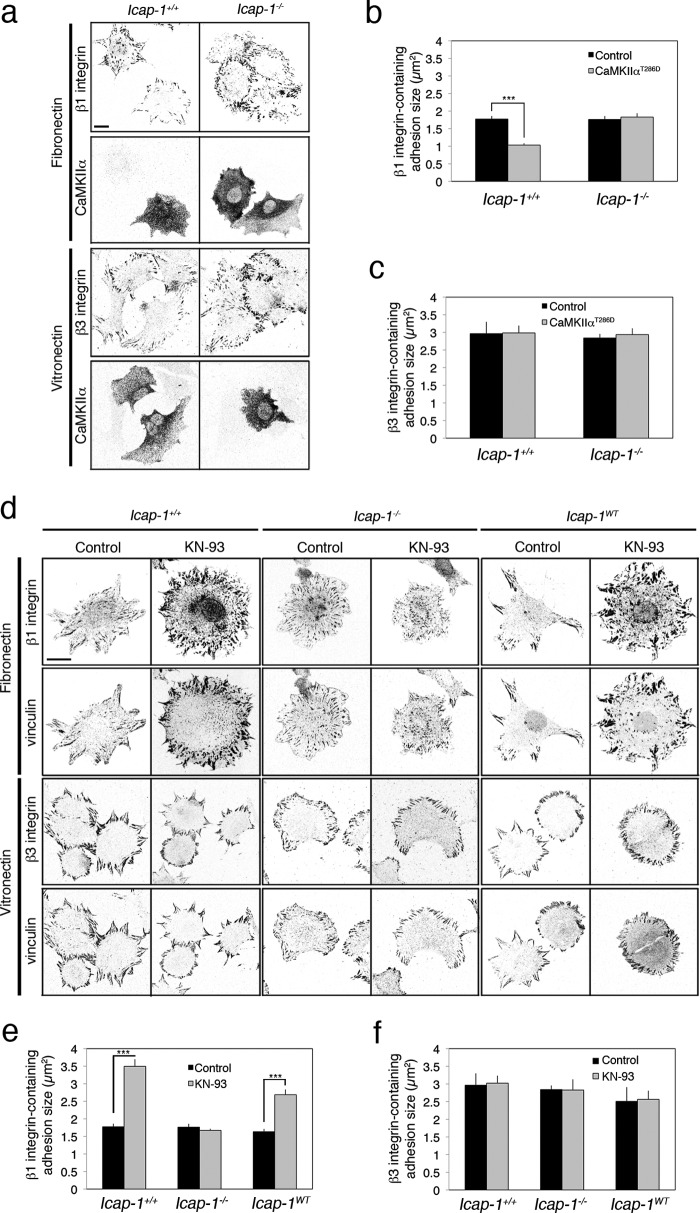
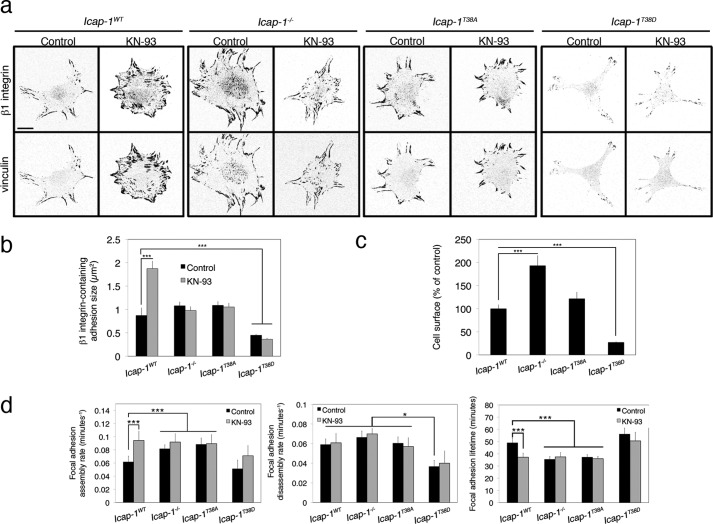
Focal adhesion turnover during cell migration is an integrated cyclic process requiring tight regulation of integrin function. Interaction of integrin with its ligand depends on its activation state, which is regulated by the direct recruitment of proteins onto the β integrin chain cytoplasmic domain. We previously reported that ICAP-1α, a specific cytoplasmic partner of β1A integrins, limits both talin and kindlin interaction with β1 integrin, thereby restraining focal adhesion assembly. Here we provide evidence that the calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII) is an important regulator of ICAP-1α for controlling focal adhesion dynamics. CaMKII directly phosphorylates ICAP-1α and disrupts an intramolecular interaction between the N- and the C-terminal domains of ICAP-1α, unmasking the PTB domain, thereby permitting ICAP-1α binding onto the β1 integrin tail. ICAP-1α direct interaction with the β1 integrin tail and the modulation of β1 integrin affinity state are required for down-regulating focal adhesion assembly. Our results point to a molecular mechanism for the phosphorylation-dependent control of ICAP-1α function by CaMKII, allowing the dynamic control of β1 integrin activation and cell adhesion.
Keywords: Adhesion; CaMKII; Focal Adhesion; ICAP-1; Integrins; Membrane Proteins; Signal Transduction.
Figures





References
-
- Hynes R. O. (2002) Integrins. Bidirectional, allosteric signaling machines. Cell 110, 673–687 - PubMed
-
- Lauffenburger D. A., Horwitz A. F. (1996) Cell migration. A physically integrated molecular process. Cell 84, 359–369 - PubMed
-
- Shimaoka M., Takagi J., Springer T. A. (2002) Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31, 485–516 - PubMed
-
- Takagi J., Petre B. M., Walz T., Springer T. A. (2002) Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
