

4-Hydroxytamoxifen induces autophagic death through K-Ras degradation
- PMID: 23722551
- PMCID: PMC3715566
- DOI: 10.1158/0008-5472.CAN-12-3765
4-Hydroxytamoxifen induces autophagic death through K-Ras degradation
Abstract
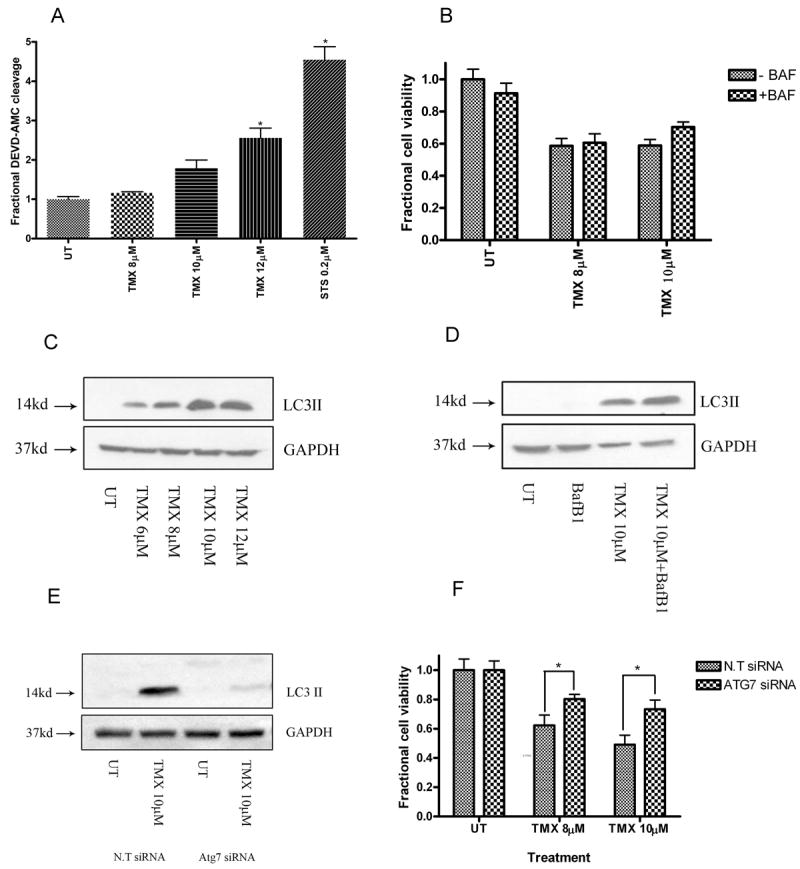
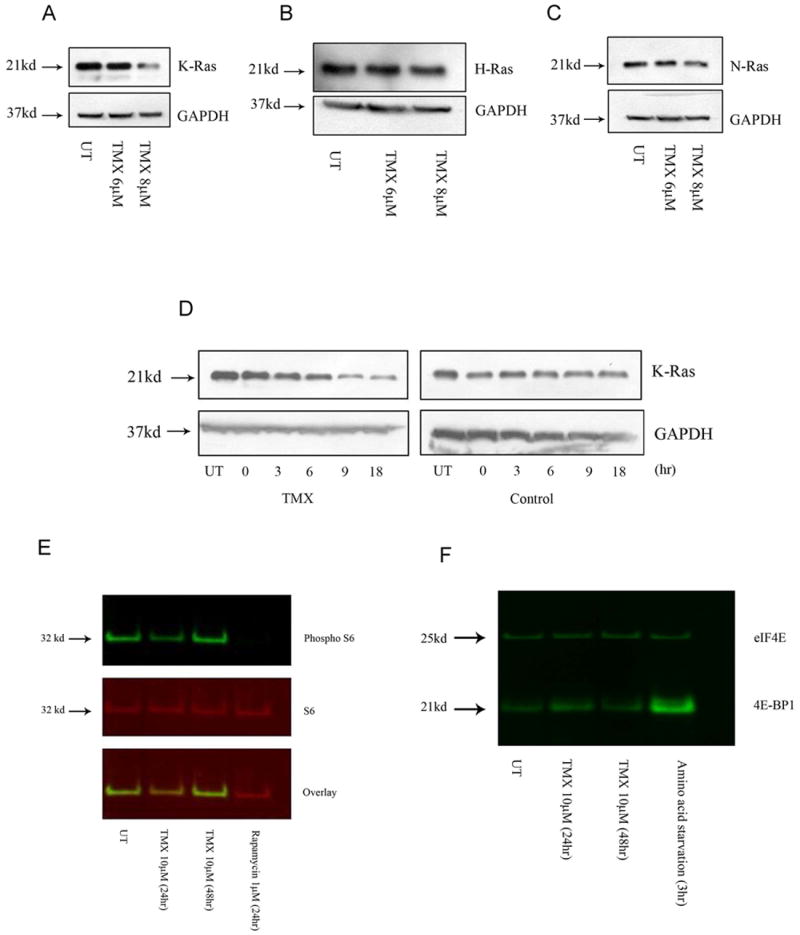
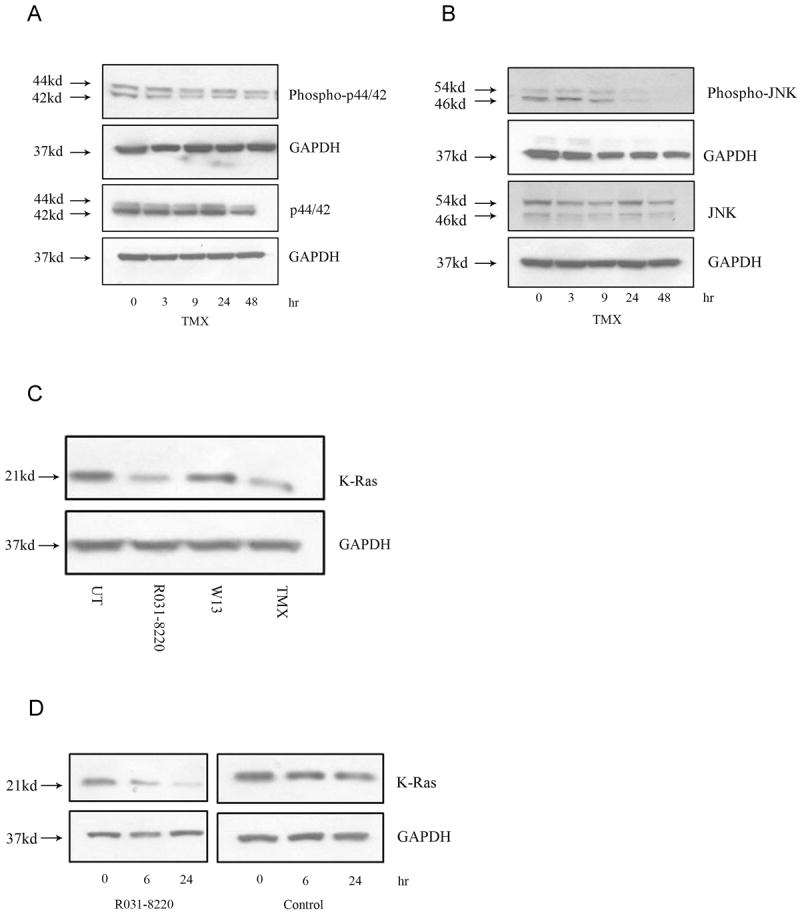
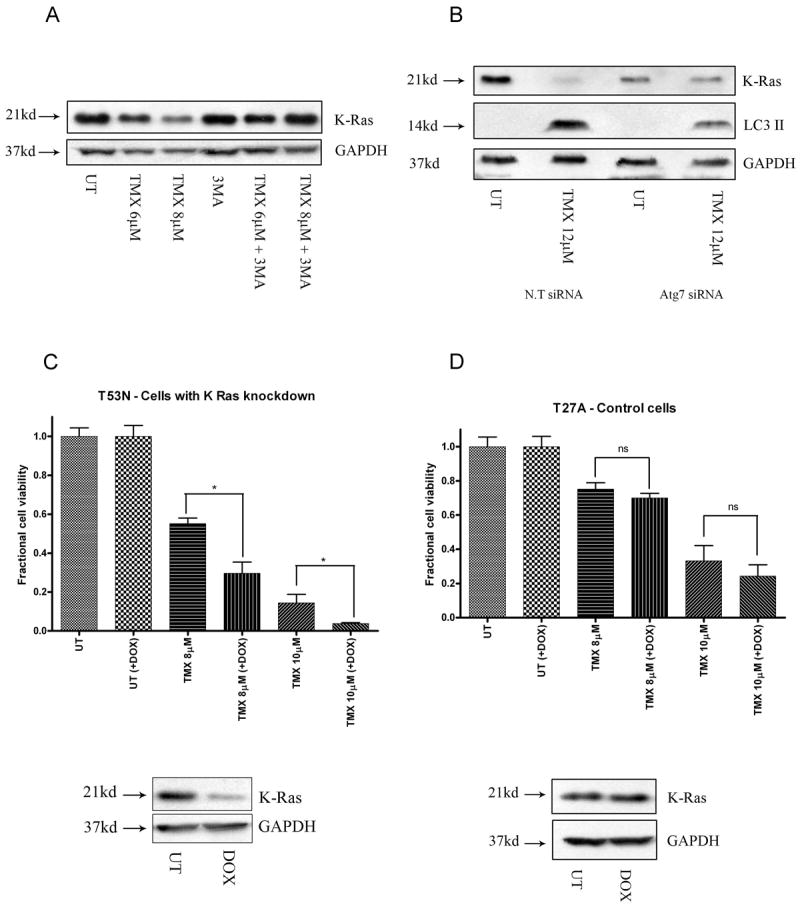
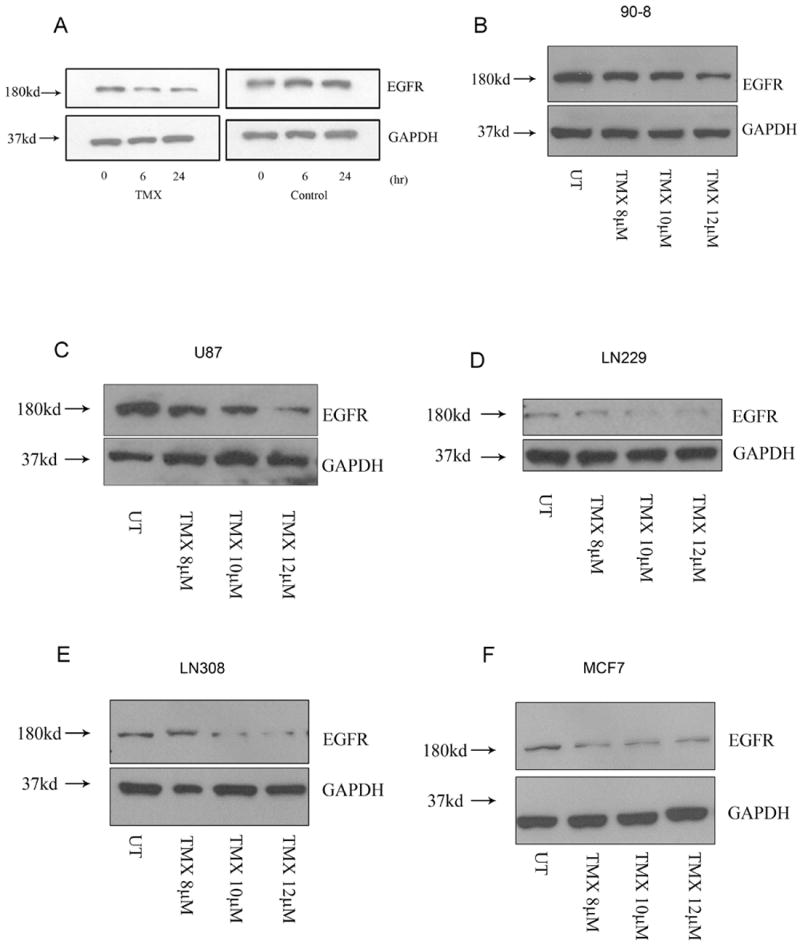
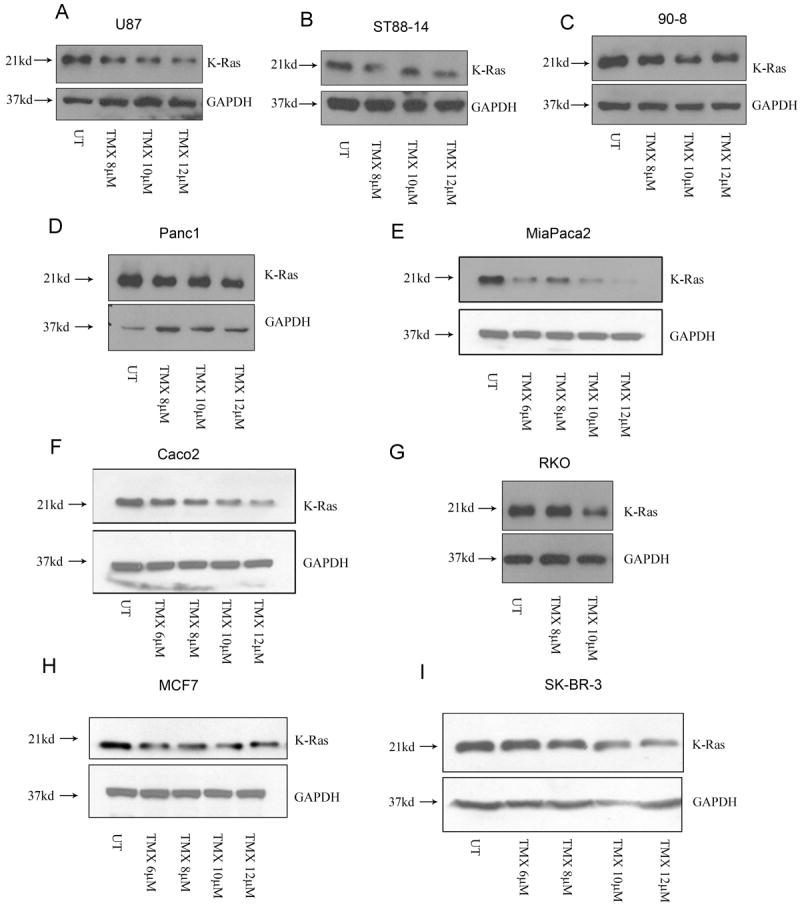
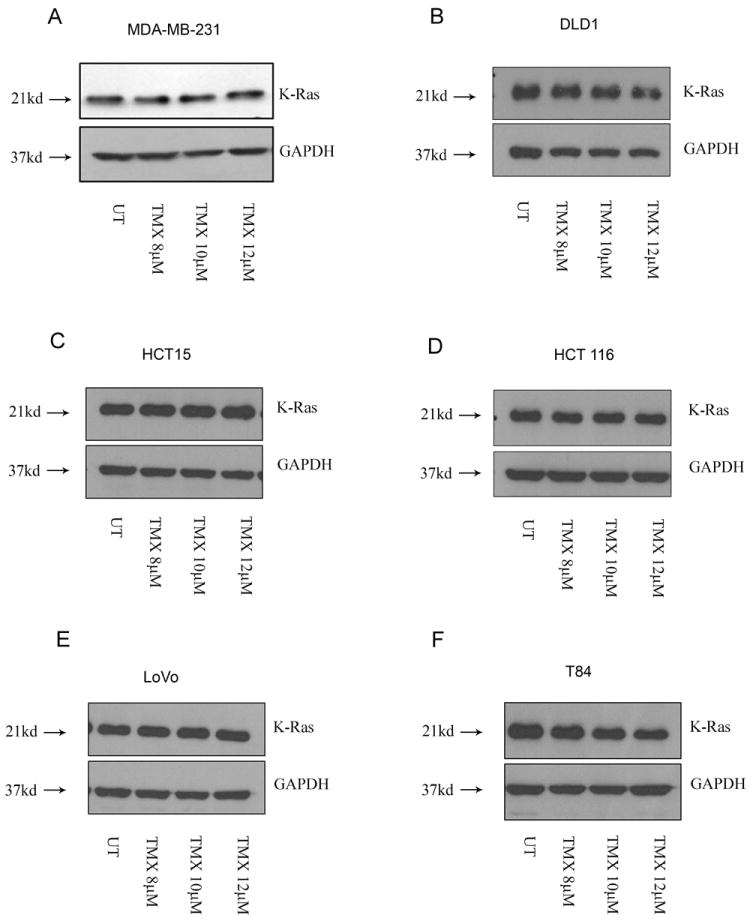
Tamoxifen is widely used to treat estrogen receptor-positive breast cancer. Recent findings that tamoxifen and its derivative 4-hydroxytamoxifen (OHT) can exert estrogen receptor-independent cytotoxic effects have prompted the initiation of clinical trials to evaluate its use in estrogen receptor-negative malignancies. For example, tamoxifen and OHT exert cytotoxic effects in malignant peripheral nerve sheath tumors (MPNST) where estrogen is not involved. In this study, we gained insights into the estrogen receptor-independent cytotoxic effects of OHT by studying how it kills MPNST cells. Although caspases were activated following OHT treatment, caspase inhibition provided no protection from OHT-induced death. Rather, OHT-induced death in MPNST cells was associated with autophagic induction and attenuated by genetic inhibition of autophagic vacuole formation. Mechanistic investigations revealed that OHT stimulated autophagic degradation of K-Ras, which is critical for survival of MPNST cells. Similarly, we found that OHT induced K-Ras degradation in breast, colon, glioma, and pancreatic cancer cells. Our findings describe a novel mechanism of autophagic death triggered by OHT in tumor cells that may be more broadly useful clinically in cancer treatment.
©2013 AACR.
Conflict of interest statement
Figures
Similar articles
-
Effect of prolonged hydroxytamoxifen treatment of MCF-7 cells on mitogen activated kinase cascade.Int J Cancer. 2002 Apr 10;98(5):698-706. doi: 10.1002/ijc.10252. Int J Cancer. 2002. PMID: 11920638
-
Estrogen receptor-dependent and estrogen receptor-independent pathways for tamoxifen and 4-hydroxytamoxifen-induced programmed cell death.J Biol Chem. 2002 Nov 22;277(47):45695-703. doi: 10.1074/jbc.M208092200. Epub 2002 Sep 19. J Biol Chem. 2002. PMID: 12244117
-
Activation of the p38 mitogen-activated protein kinase pathway by estrogen or by 4-hydroxytamoxifen is coupled to estrogen receptor-induced apoptosis.J Biol Chem. 2000 Jan 7;275(1):479-86. doi: 10.1074/jbc.275.1.479. J Biol Chem. 2000. PMID: 10617642
-
4-Hydroxytamoxifen inhibits proliferation of multiple myeloma cells in vitro through down-regulation of c-Myc, up-regulation of p27Kip1, and modulation of Bcl-2 family members.Clin Cancer Res. 2005 Mar 15;11(6):2345-54. doi: 10.1158/1078-0432.CCR-04-1668. Clin Cancer Res. 2005. PMID: 15788686
-
c-Jun activation is required for 4-hydroxytamoxifen-induced cell death in breast cancer cells.Oncogene. 2010 Feb 18;29(7):978-91. doi: 10.1038/onc.2009.400. Epub 2009 Nov 23. Oncogene. 2010. PMID: 19935718
Cited by
-
Potent and broad anticancer activities of leaf extracts from Melia azedarach L. of the subtropical Okinawa islands.Am J Cancer Res. 2020 Feb 1;10(2):581-594. eCollection 2020. Am J Cancer Res. 2020. PMID: 32195029 Free PMC article.
-
Autophagy manipulation as a strategy for efficient anticancer therapies: possible consequences.J Exp Clin Cancer Res. 2019 Jun 14;38(1):262. doi: 10.1186/s13046-019-1275-z. J Exp Clin Cancer Res. 2019. PMID: 31200739 Free PMC article. Review.
-
Autophagy induction causes a synthetic lethal sensitization to ribonucleotide reductase inhibition in breast cancer cells.Oncotarget. 2016 Jan 12;7(2):1984-99. doi: 10.18632/oncotarget.6539. Oncotarget. 2016. PMID: 26675256 Free PMC article.
-
Mincle-Mediated Neutrophil Extracellular Trap Formation by Regulation of Autophagy.J Infect Dis. 2017 Apr 1;215(7):1040-1048. doi: 10.1093/infdis/jix072. J Infect Dis. 2017. PMID: 28186242 Free PMC article.
-
Chloroquine and hydroxychloroquine for cancer therapy.Mol Cell Oncol. 2014 Jul 15;1(1):e29911. doi: 10.4161/mco.29911. eCollection 2014. Mol Cell Oncol. 2014. PMID: 27308318 Free PMC article. Review.
References
-
- Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997 Jun;11(6):657–66. - PubMed
-
- Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001 Dec;6(6):469–77. - PubMed
-
- Zhang W, Couldwell WT, Song H, Takano T, Lin JH, Nedergaard M. Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells. Cancer Res. 2000 Oct 1;60(19):5395–400. - PubMed
-
- Gundimeda U, Chen ZH, Gopalakrishna R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor-negative breast cancer cells. J Biol Chem. 1996 Jun 7;271(23):13504–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
