

Modulation of lipid kinase PI4KIIα activity and lipid raft association of presenilin 1 underlies γ-secretase inhibition by ginsenoside (20S)-Rg3
- PMID: 23723072
- PMCID: PMC3774358
- DOI: 10.1074/jbc.M112.445734
Modulation of lipid kinase PI4KIIα activity and lipid raft association of presenilin 1 underlies γ-secretase inhibition by ginsenoside (20S)-Rg3
Abstract
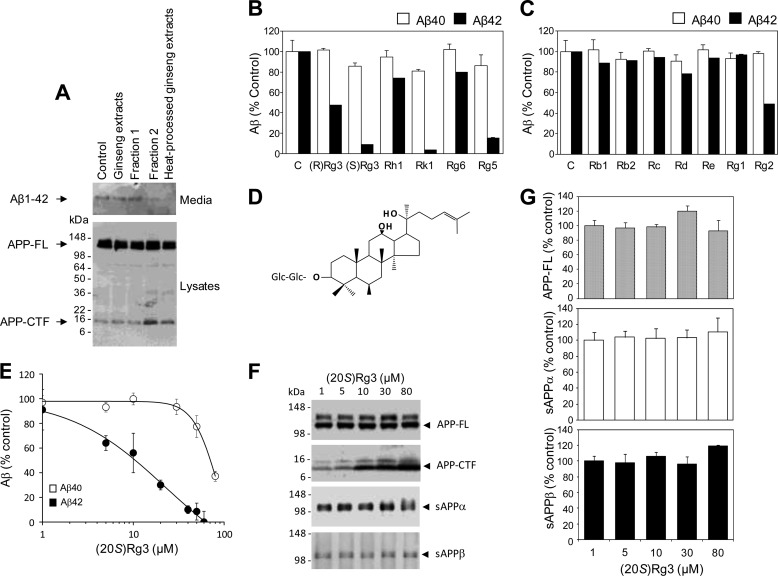
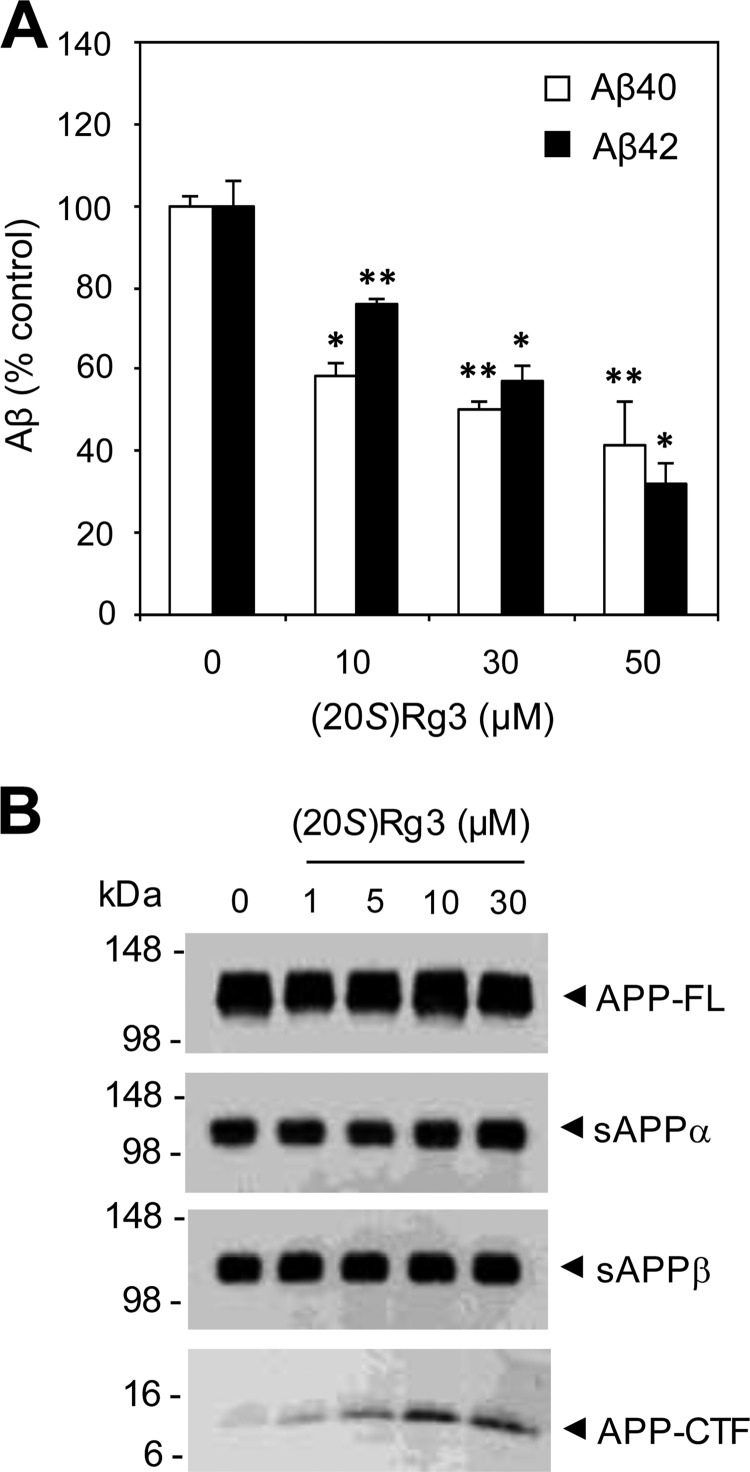
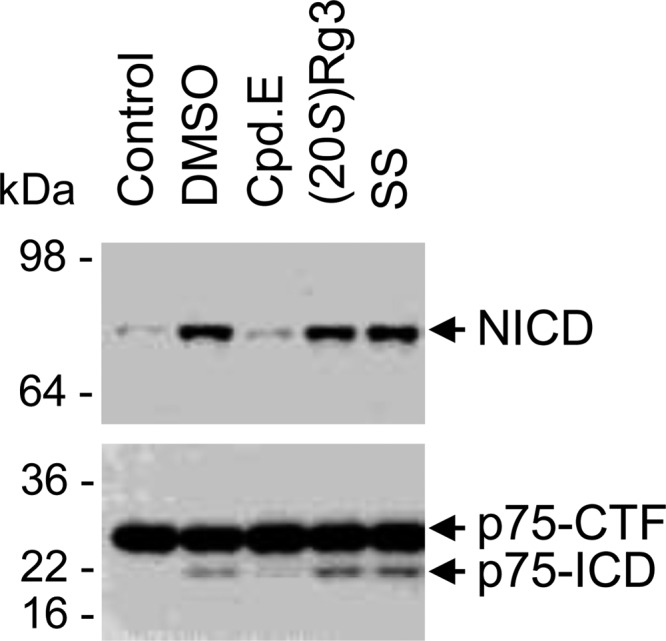
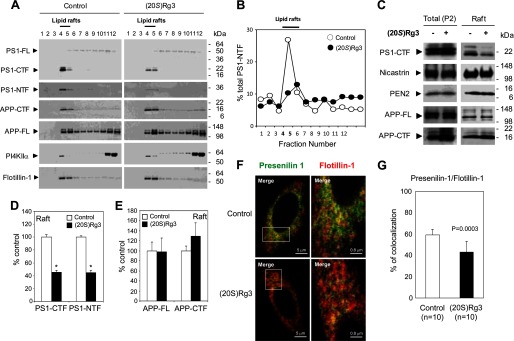
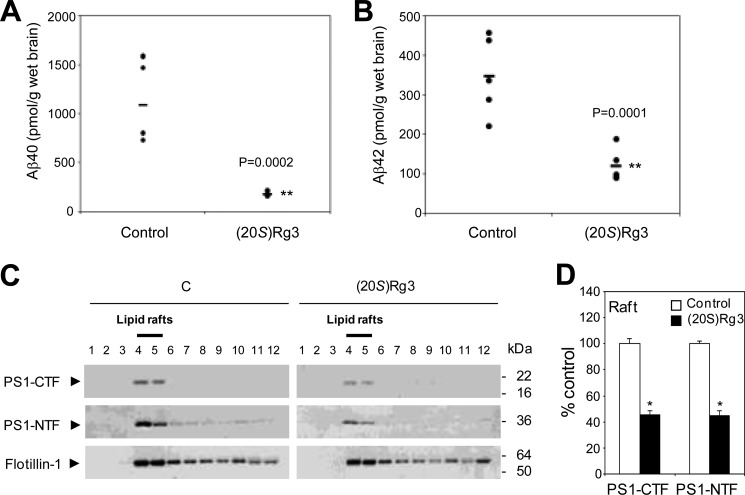
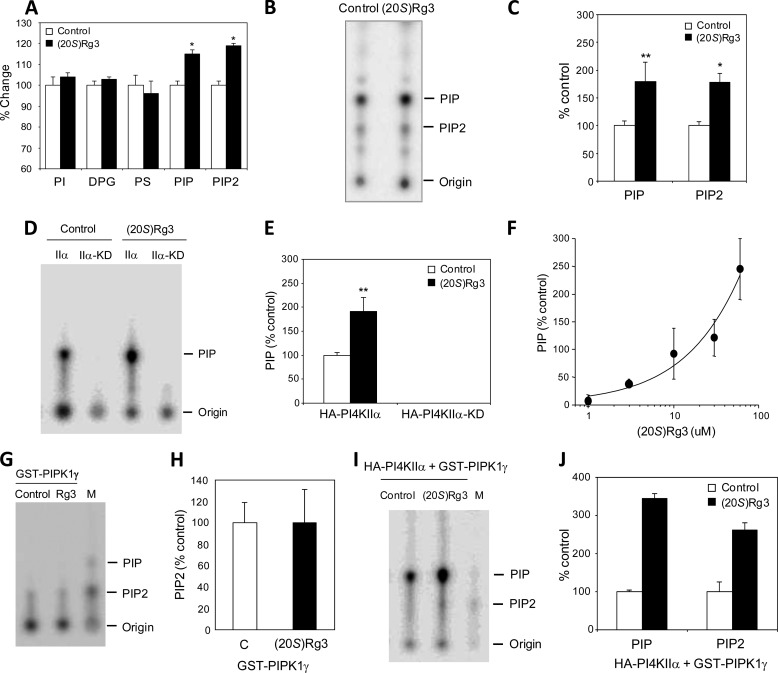
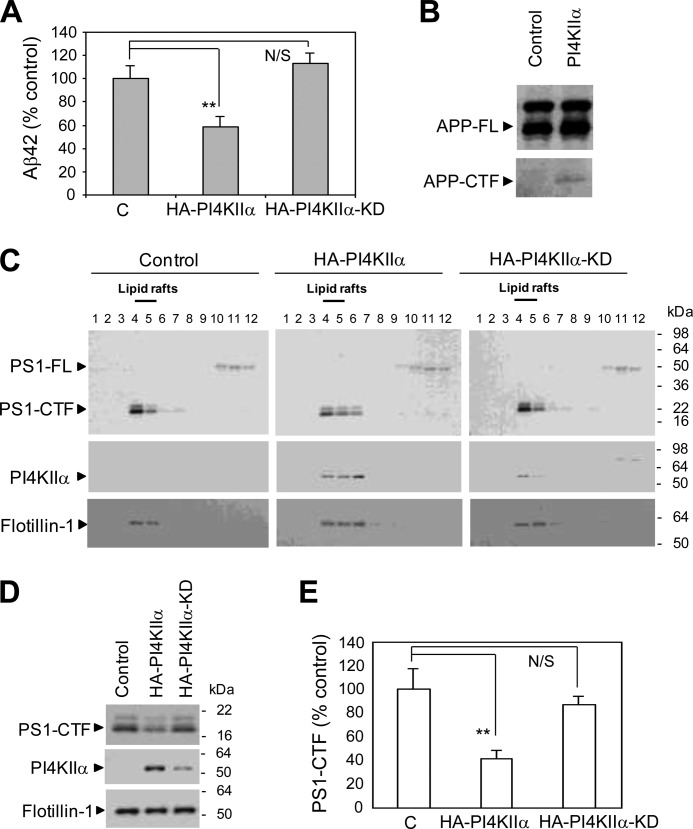
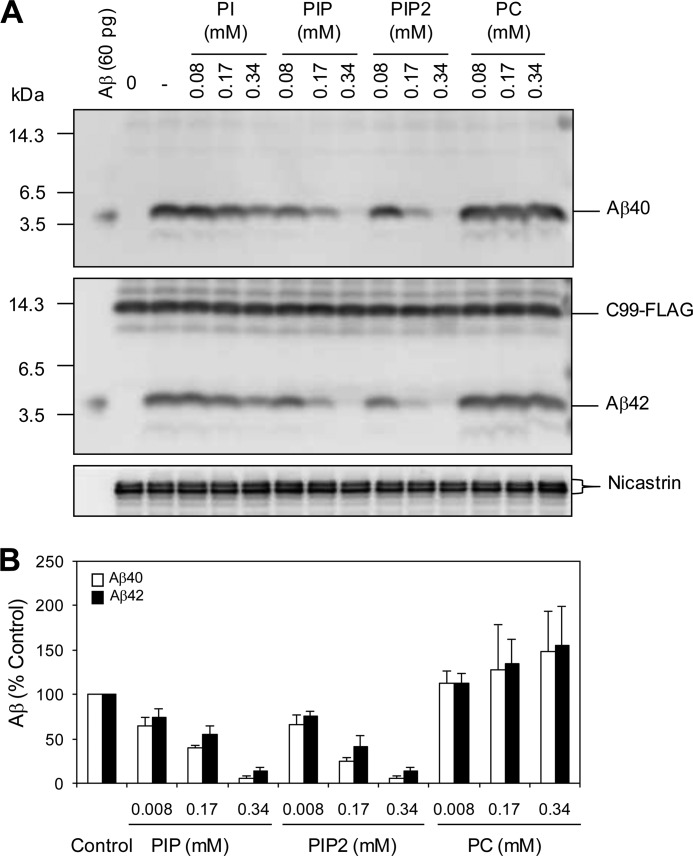
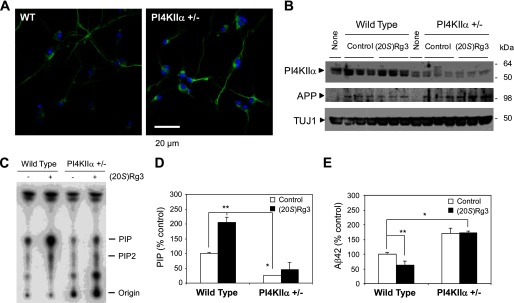
Amyloid β-peptide (Aβ) pathology is an invariant feature of Alzheimer disease, preceding any detectable clinical symptoms by more than a decade. To this end, we seek to identify agents that can reduce Aβ levels in the brain via novel mechanisms. We found that (20S)-Rg3, a triterpene natural compound known as ginsenoside, reduced Aβ levels in cultured primary neurons and in the brains of a mouse model of Alzheimer disease. The (20S)-Rg3 treatment induced a decrease in the association of presenilin 1 (PS1) fragments with lipid rafts where catalytic components of the γ-secretase complex are enriched. The Aβ-lowering activity of (20S)-Rg3 directly correlated with increased activity of phosphatidylinositol 4-kinase IIα (PI4KIIα), a lipid kinase that mediates the rate-limiting step in phosphatidylinositol 4,5-bisphosphate synthesis. PI4KIIα overexpression recapitulated the effects of (20S)-Rg3, whereas reduced expression of PI4KIIα abolished the Aβ-reducing activity of (20S)-Rg3 in neurons. Our results substantiate an important role for PI4KIIα and phosphoinositide modulation in γ-secretase activity and Aβ biogenesis.
Keywords: Alzheimer Disease; Lipid Raft; Lipids; Natural Products; Phosphatidylinositol; Presenilin.
Figures
Similar articles
-
TASTPM mice expressing amyloid precursor protein and presenilin-1 mutant transgenes are sensitive to γ-secretase modulation and amyloid-β₄₂ lowering by GSM-10h.Neurodegener Dis. 2011;8(1-2):15-24. doi: 10.1159/000313903. Epub 2010 Aug 4. Neurodegener Dis. 2011. PMID: 20689247
-
Mild cholesterol depletion reduces amyloid-beta production by impairing APP trafficking to the cell surface.J Neurochem. 2009 Jul;110(1):220-30. doi: 10.1111/j.1471-4159.2009.06126.x. Epub 2009 Apr 27. J Neurochem. 2009. PMID: 19457132 Free PMC article.
-
Identification of novel γ-secretase-associated proteins in detergent-resistant membranes from brain.J Biol Chem. 2012 Apr 6;287(15):11991-2005. doi: 10.1074/jbc.M111.246074. Epub 2012 Feb 7. J Biol Chem. 2012. PMID: 22315232 Free PMC article.
-
Role of presenilin in gamma-secretase cleavage of amyloid precursor protein.Exp Gerontol. 2000 Jul;35(4):453-60. doi: 10.1016/s0531-5565(00)00111-x. Exp Gerontol. 2000. PMID: 10959033 Review.
-
Proton myo-inositol cotransporter is a novel γ-secretase associated protein that regulates Aβ production without affecting Notch cleavage.FEBS J. 2015 Sep;282(17):3438-51. doi: 10.1111/febs.13353. Epub 2015 Jul 14. FEBS J. 2015. PMID: 26094765 Review.
Cited by
-
Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate.J Cell Biol. 2015 May 11;209(3):435-52. doi: 10.1083/jcb.201409082. Epub 2015 May 4. J Cell Biol. 2015. PMID: 25940347 Free PMC article.
-
The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer's Disease and Other Neurodegenerative Disorders.Mol Neurobiol. 2019 Aug;56(8):5436-5455. doi: 10.1007/s12035-018-1448-3. Epub 2019 Jan 5. Mol Neurobiol. 2019. PMID: 30612333 Free PMC article. Review.
-
The Use of Nutraceuticals to Counteract Atherosclerosis: The Role of the Notch Pathway.Oxid Med Cell Longev. 2019 May 2;2019:5470470. doi: 10.1155/2019/5470470. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31915510 Free PMC article. Review.
-
Therapeutic Candidates for Alzheimer's Disease: Saponins.Int J Mol Sci. 2023 Jun 22;24(13):10505. doi: 10.3390/ijms241310505. Int J Mol Sci. 2023. PMID: 37445682 Free PMC article. Review.
-
Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains.Molecules. 2020 Nov 24;25(23):5490. doi: 10.3390/molecules25235490. Molecules. 2020. PMID: 33255194 Free PMC article.
References
-
- Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 - PubMed
-
- Tanzi R. E., Bertram L. (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555 - PubMed
-
- Mangialasche F., Solomon A., Winblad B., Mecocci P., Kivipelto M. (2010) Alzheimer's disease, clinical trials and drug development. Lancet Neurol. 9, 702–716 - PubMed
-
- Bateman R. J., Xiong C., Benzinger T. L., Fagan A. M., Goate A., Fox N. C., Marcus D. S., Cairns N. J., Xie X., Blazey T. M., Holtzman D. M., Santacruz A., Buckles V., Oliver A., Moulder K., Aisen P. S., Ghetti B., Klunk W. E., McDade E., Martins R. N., Masters C. L., Mayeux R., Ringman J. M., Rossor M. N., Schofield P. R., Sperling R. A., Salloway S., Morris J. C.; Dominantly Inherited Alzheimer Network (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
