

The DcpS inhibitor RG3039 improves motor function in SMA mice
- PMID: 23727836
- PMCID: PMC3781637
- DOI: 10.1093/hmg/ddt257
The DcpS inhibitor RG3039 improves motor function in SMA mice
Abstract
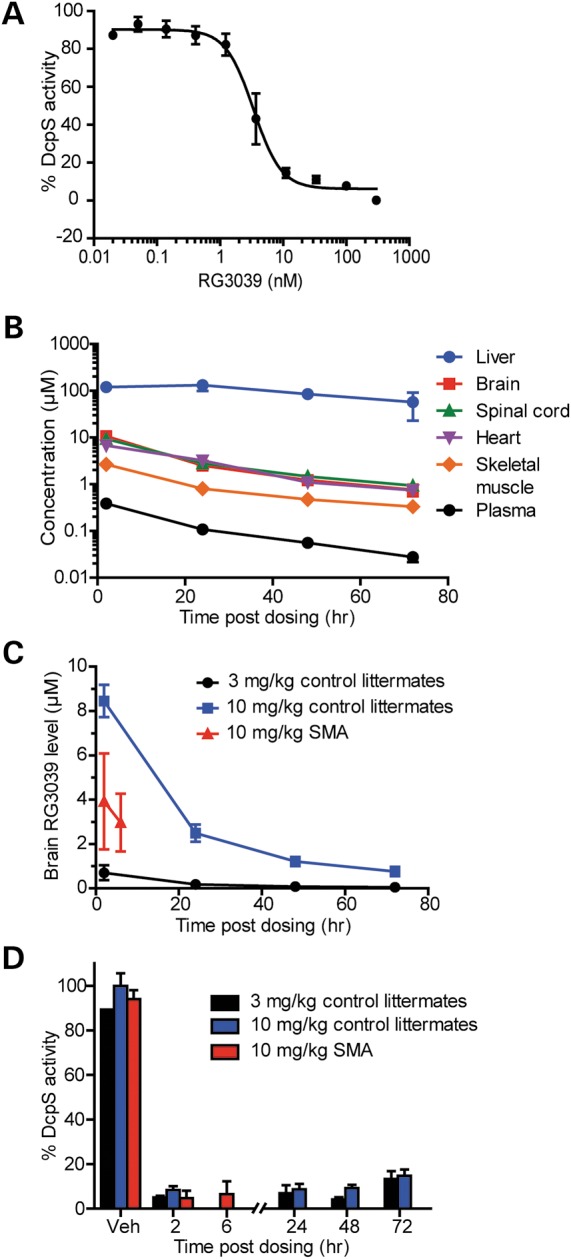
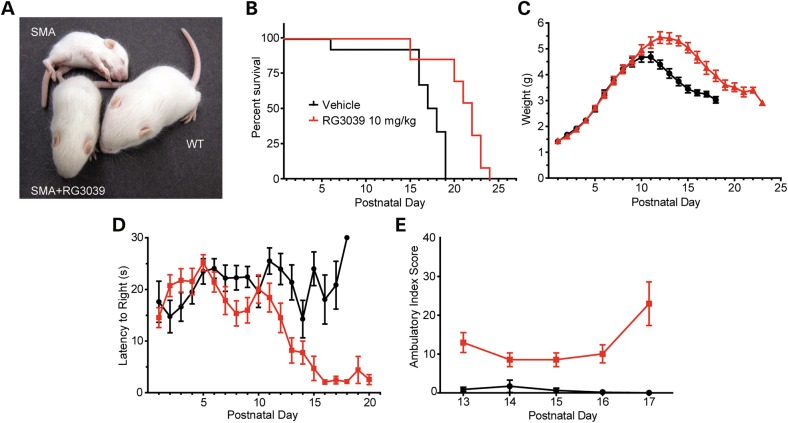
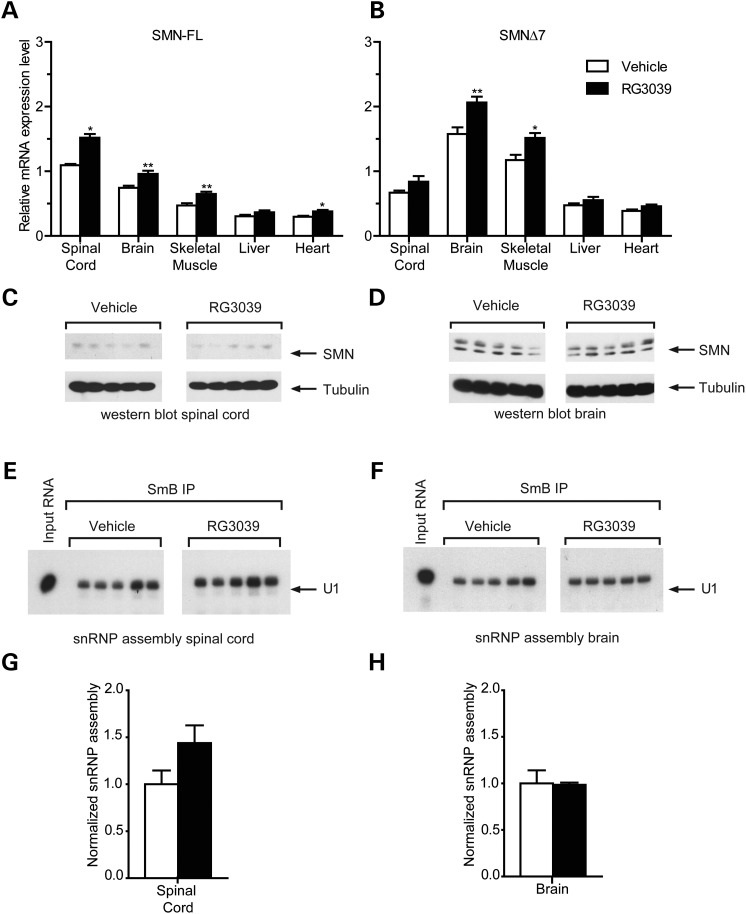
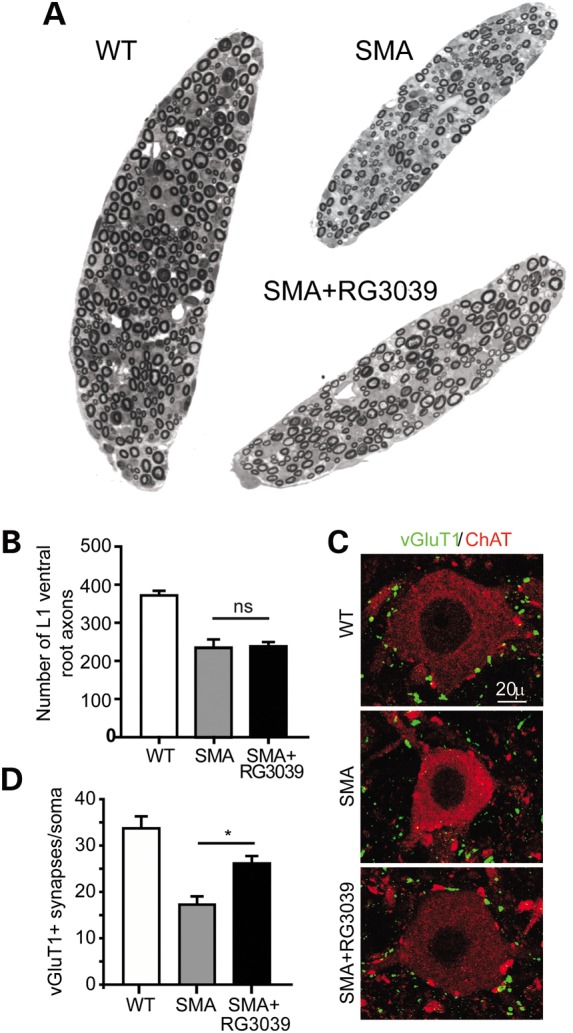
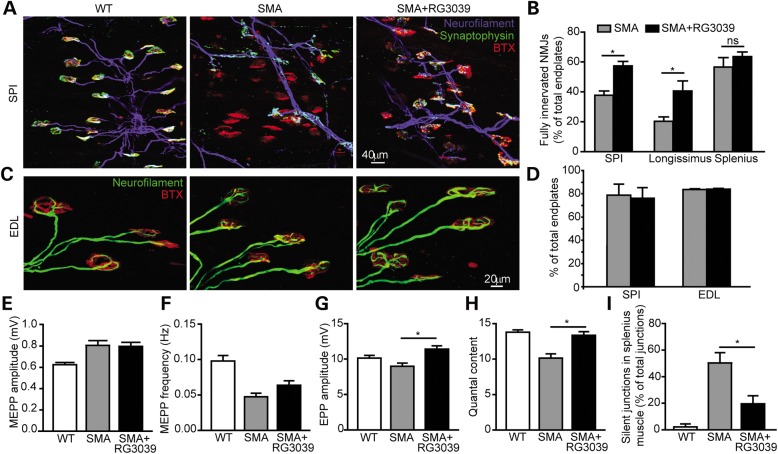
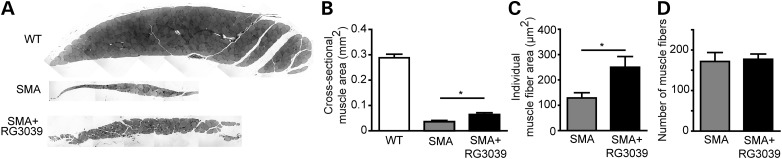
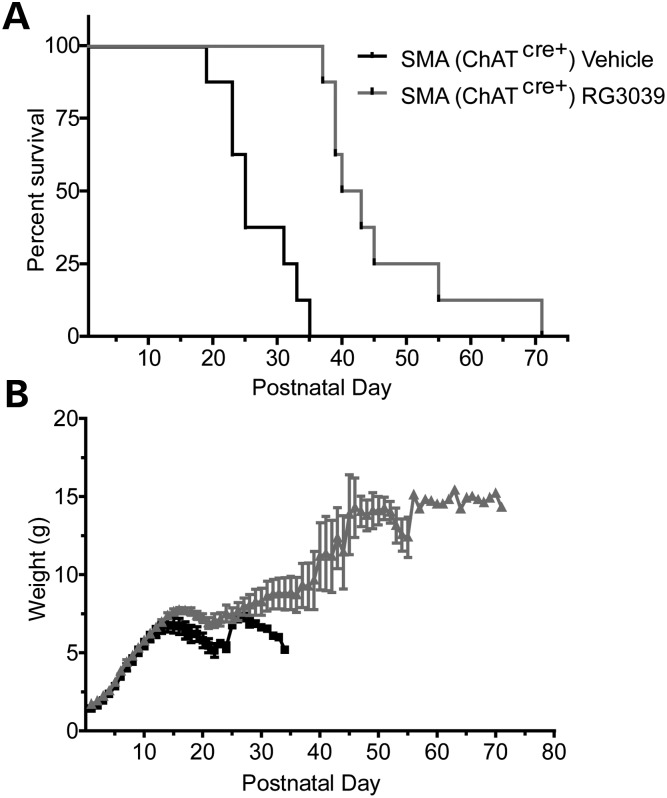
Spinal muscular atrophy (SMA) is caused by mutations of the survival motor neuron 1 (SMN1) gene, retention of the survival motor neuron 2 (SMN2) gene and insufficient expression of full-length survival motor neuron (SMN) protein. Quinazolines increase SMN2 promoter activity and inhibit the ribonucleic acid scavenger enzyme DcpS. The quinazoline derivative RG3039 has advanced to early phase clinical trials. In preparation for efficacy studies in SMA patients, we investigated the effects of RG3039 in severe SMA mice. Here, we show that RG3039 distributed to central nervous system tissues where it robustly inhibited DcpS enzyme activity, but minimally activated SMN expression or the assembly of small nuclear ribonucleoproteins. Nonetheless, treated SMA mice showed a dose-dependent increase in survival, weight and motor function. This was associated with improved motor neuron somal and neuromuscular junction synaptic innervation and function and increased muscle size. RG3039 also enhanced survival of conditional SMA mice in which SMN had been genetically restored to motor neurons. As this systemically delivered drug may have therapeutic benefits that extend beyond motor neurons, it could act additively with SMN-restoring therapies delivered directly to the central nervous system such as antisense oligonucleotides or gene therapy.
Figures
References
-
- Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. - PubMed
-
- Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
