

C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation
- PMID: 23728178
- PMCID: PMC3668852
- DOI: 10.1172/JCI68280
C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation
Abstract
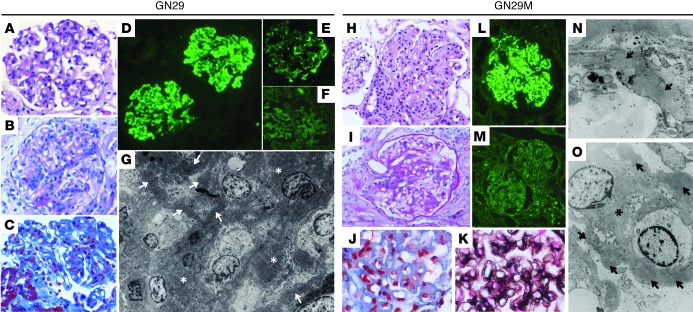
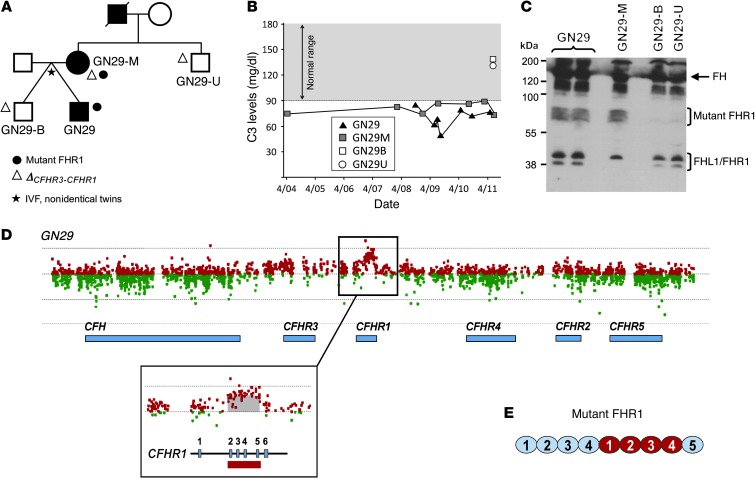
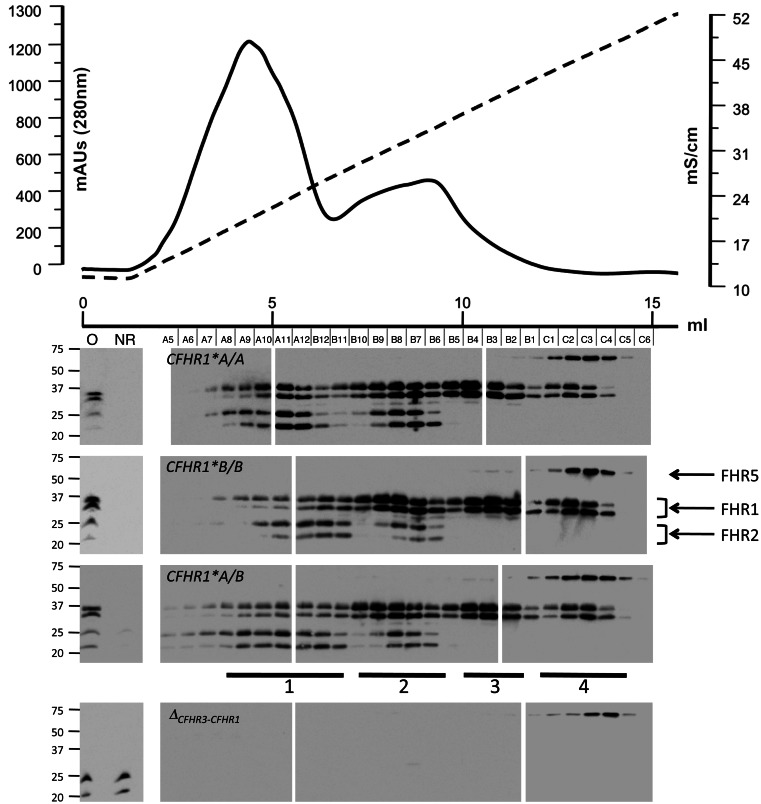
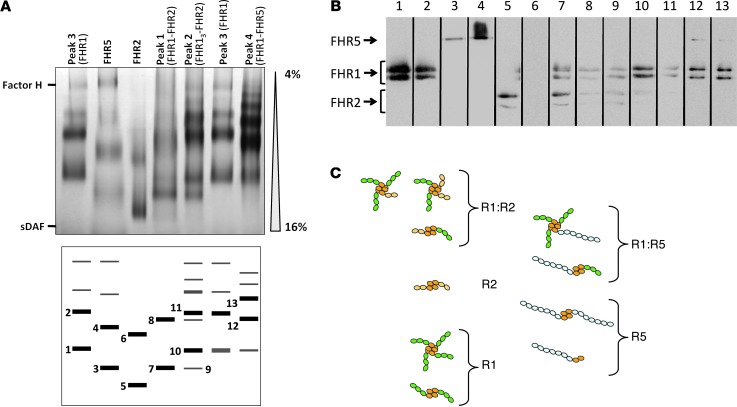
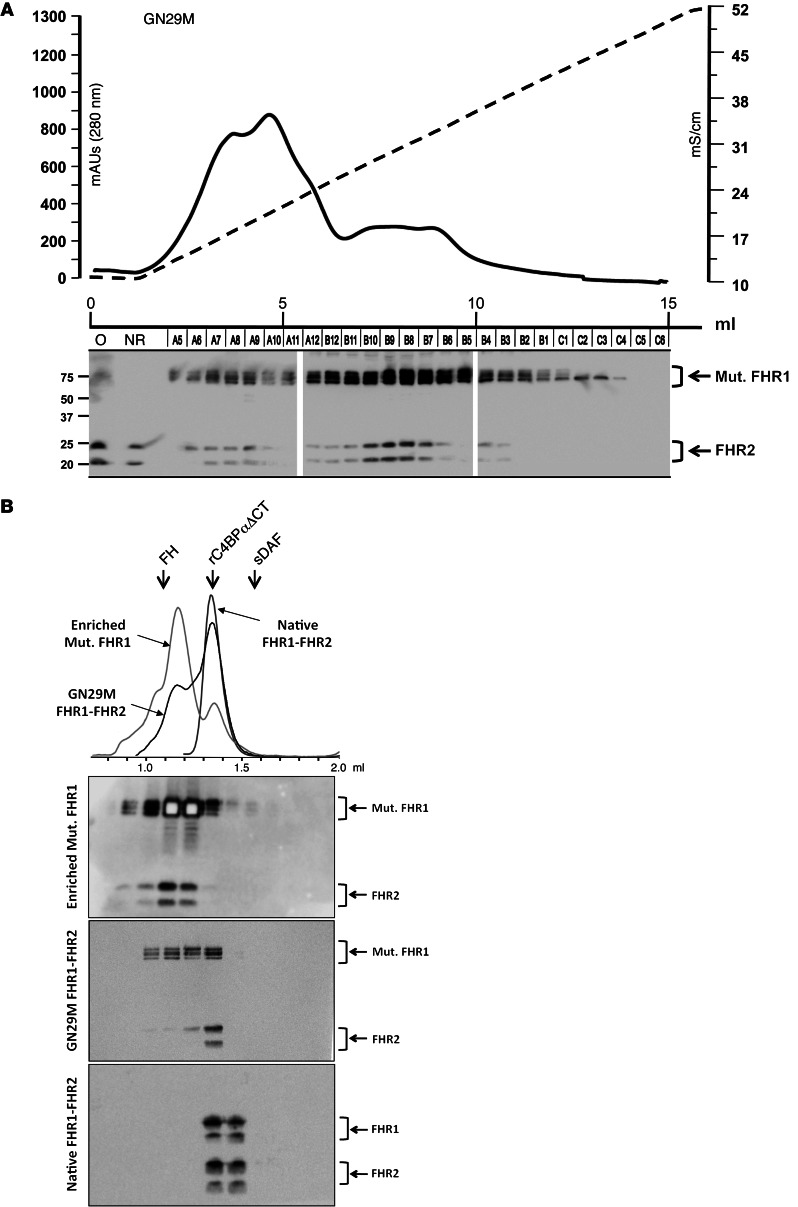
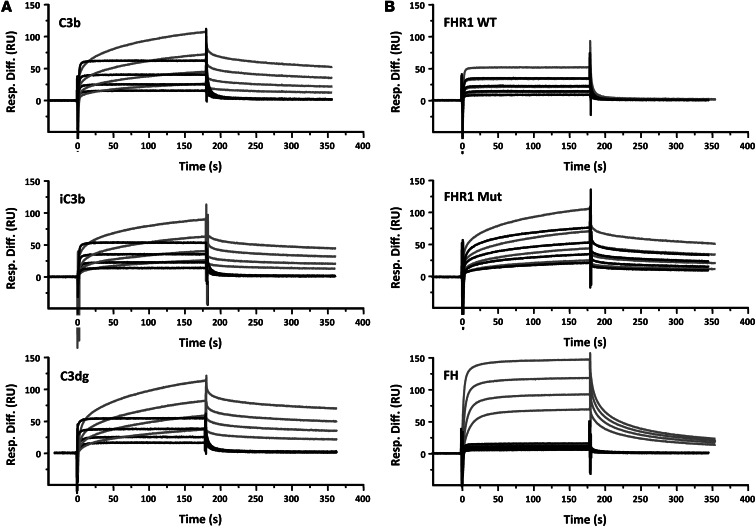
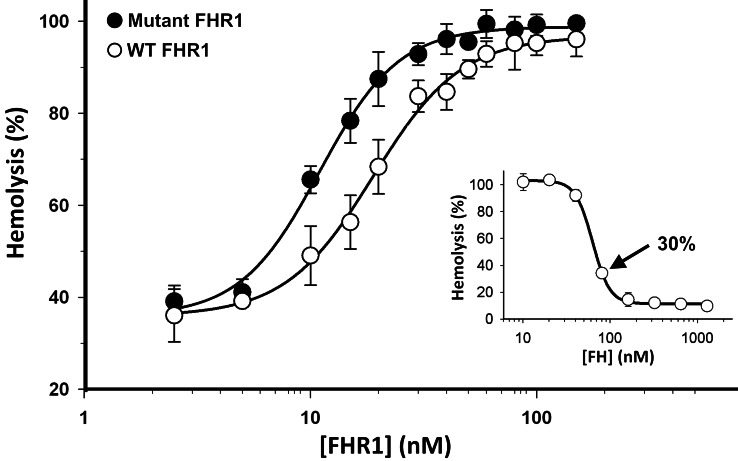
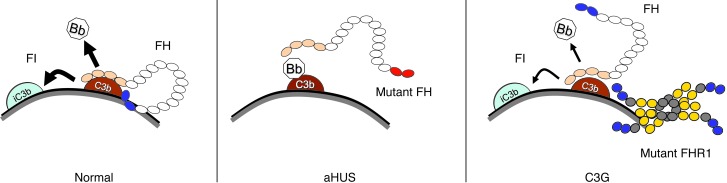
C3 glomerulopathies (C3G) are a group of severe renal diseases with distinct patterns of glomerular inflammation and C3 deposition caused by complement dysregulation. Here we report the identification of a familial C3G-associated genomic mutation in the gene complement factor H–related 1 (CFHR1), which encodes FHR1. The mutation resulted in the duplication of the N-terminal short consensus repeats (SCRs) that are conserved in FHR2 and FHR5. We determined that native FHR1, FHR2, and FHR5 circulate in plasma as homo- and hetero-oligomeric complexes, the formation of which is likely mediated by the conserved N-terminal domain. In mutant FHR1, duplication of the N-terminal domain resulted in the formation of unusually large multimeric FHR complexes that exhibited increased avidity for the FHR1 ligands C3b, iC3b, and C3dg and enhanced competition with complement factor H (FH) in surface plasmon resonance (SPR) studies and hemolytic assays. These data revealed that FHR1, FHR2, and FHR5 organize a combinatorial repertoire of oligomeric complexes and demonstrated that changes in FHR oligomerization influence the regulation of complement activation. In summary, our identification and characterization of a unique CFHR1 mutation provides insights into the biology of the FHRs and contributes to our understanding of the pathogenic mechanisms underlying C3G.
Figures

Comment in
-
Human C3 glomerulopathy provides unique insights into complement factor H-related protein function.J Clin Invest. 2013 Jun;123(6):2357-60. doi: 10.1172/JCI69684. J Clin Invest. 2013. PMID: 23728171 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
