

Two-photon excitation microscopy for the study of living cells and tissues
- PMID: 23728746
- PMCID: PMC4004770
- DOI: 10.1002/0471143030.cb0411s59
Two-photon excitation microscopy for the study of living cells and tissues
Abstract
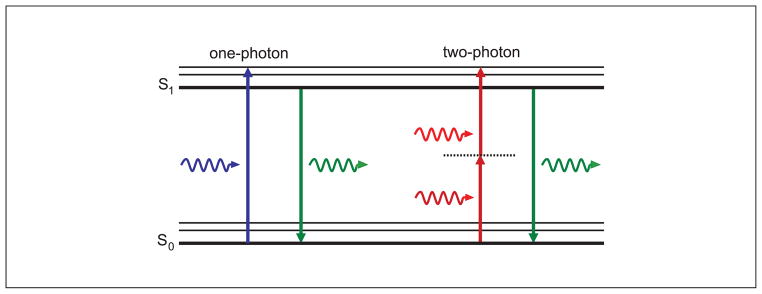
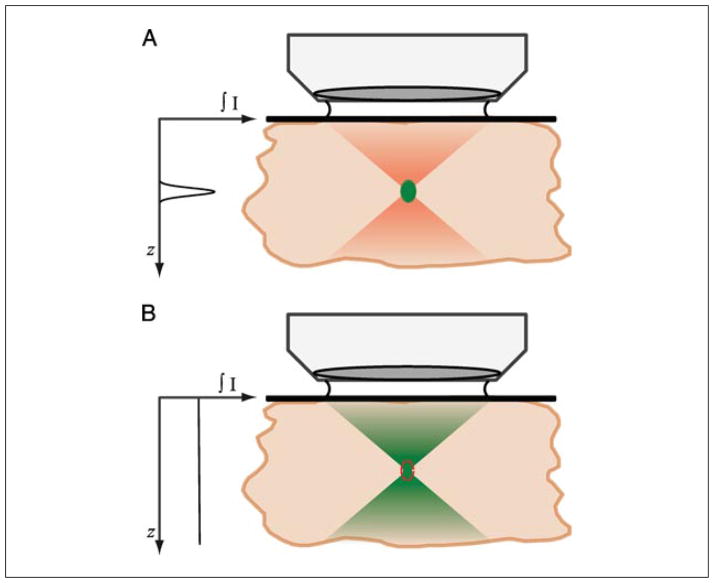
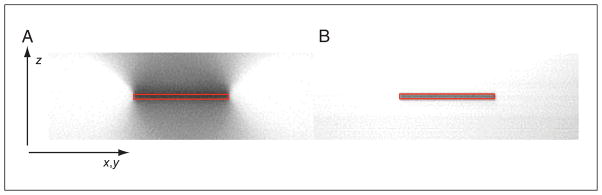
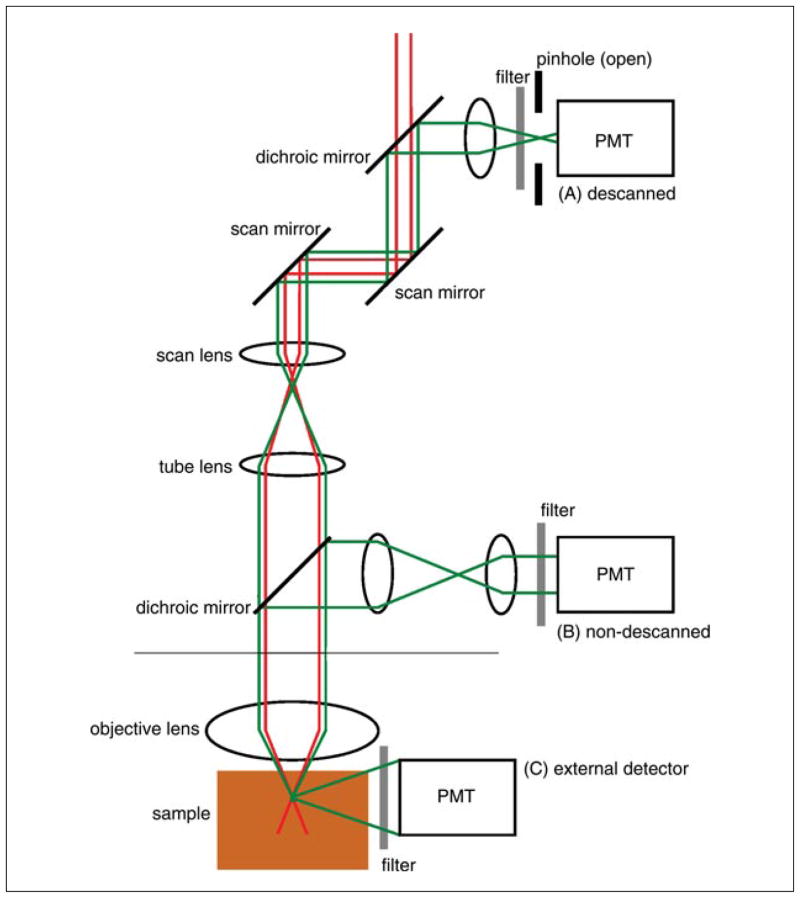
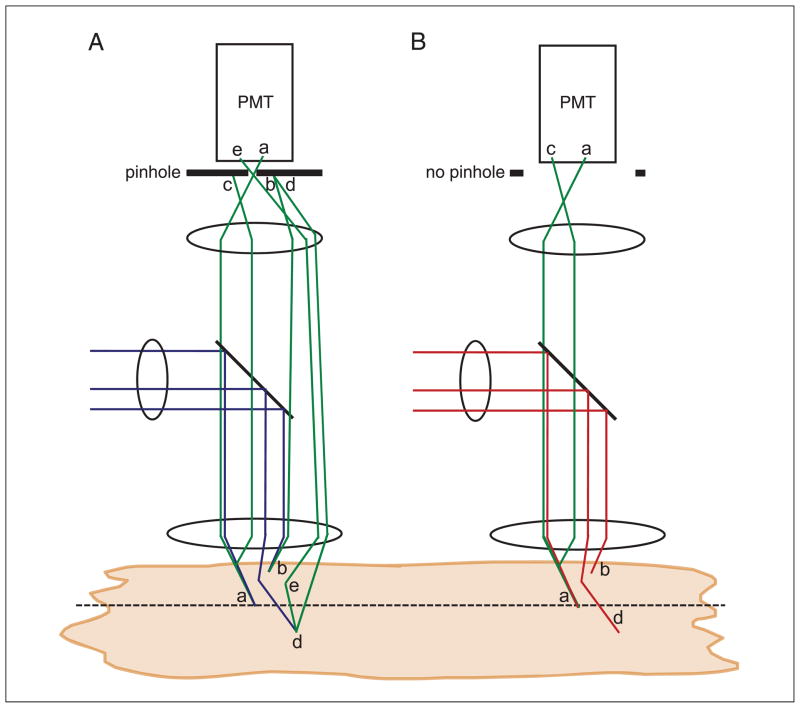
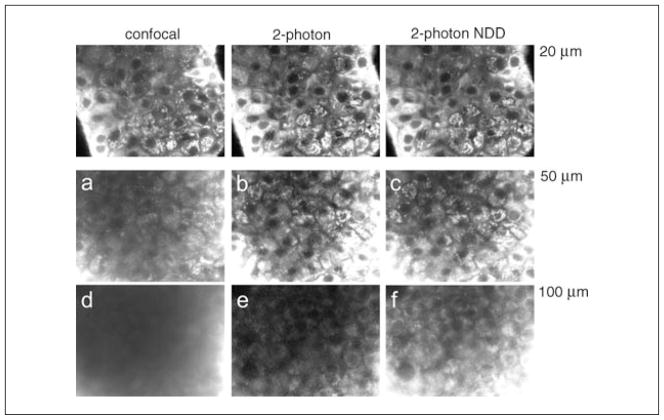
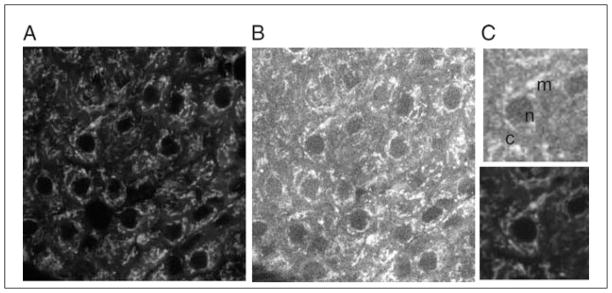
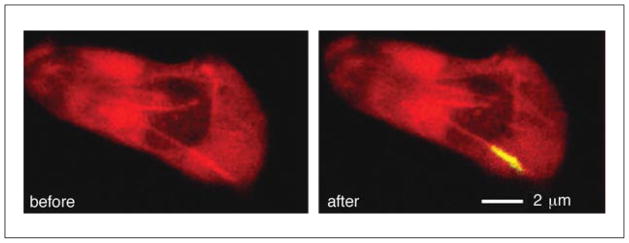
Two-photon excitation microscopy is an alternative to confocal microscopy that provides advantages for three-dimensional and deep tissue imaging. This unit will describe the basic physical principles behind two-photon excitation and discuss the advantages and limitations of its use in laser-scanning microscopy. The principal advantages of two-photon microscopy are reduced phototoxicity, increased imaging depth, and the ability to initiate highly localized photochemistry in thick samples. Practical considerations for the application of two-photon microscopy will then be discussed, including recent technological advances. This unit will conclude with some recent applications of two-photon microscopy that highlight the key advantages over confocal microscopy and the types of experiments which would benefit most from its application.
© 2013 by John Wiley & Sons, Inc.
Figures
References
-
- Albota MA, Xu C, Webb WW. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl Optics. 1998;37:7352–7356. - PubMed
-
- Beaurepaire E, Oheim M, Mertz J. Ultra-deep two-photon fluorescence excitation in turbid media. Opt Comm. 2001;188:25–29.
-
- Bennett BD, Jetton TL, Ying G, Magnuson MA, Piston DW. Quantitative sub-cellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem. 1996;271:3647–3651. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
