

BeEP Server: Using evolutionary information for quality assessment of protein structure models
- PMID: 23729471
- PMCID: PMC3692104
- DOI: 10.1093/nar/gkt453
BeEP Server: Using evolutionary information for quality assessment of protein structure models
Abstract
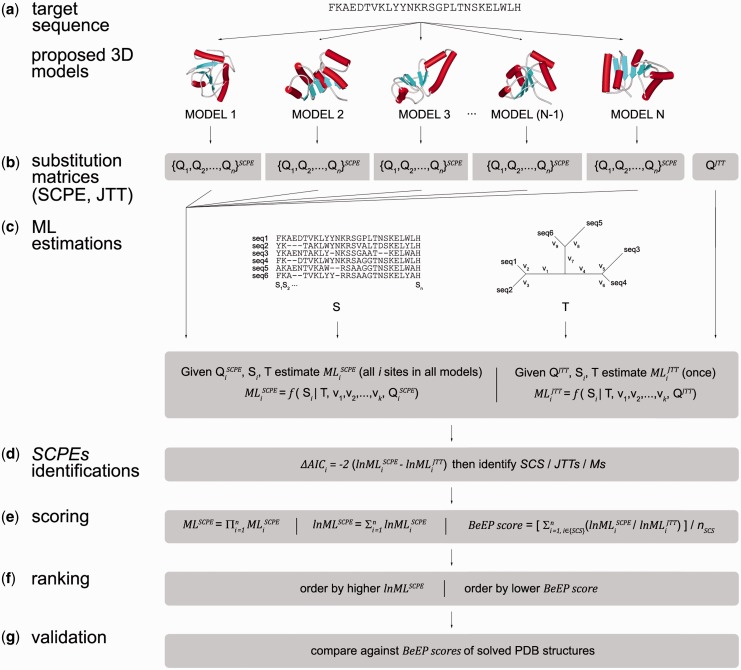
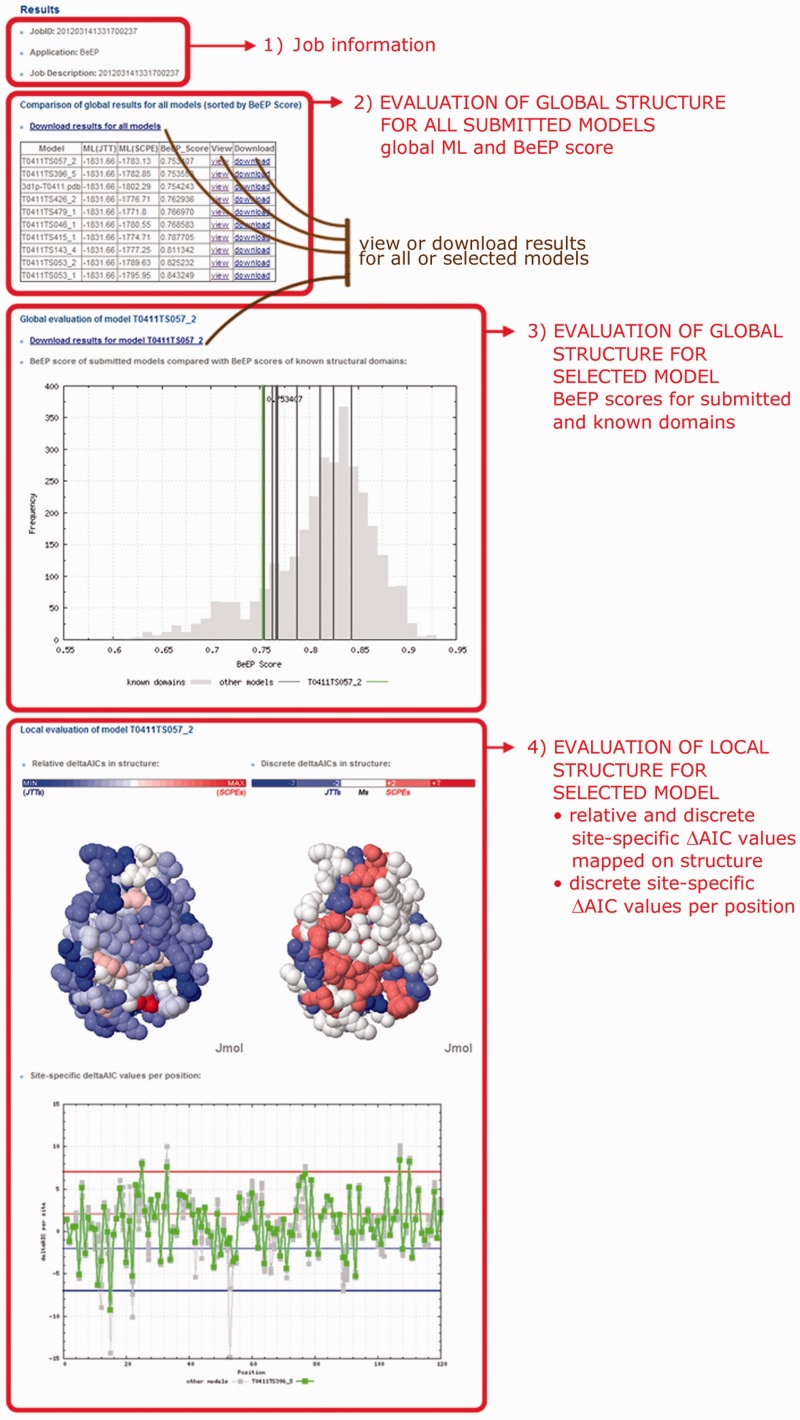
The BeEP Server (http://www.embnet.qb.fcen.uba.ar/embnet/beep.php) is an online resource aimed to help in the endgame of protein structure prediction. It is able to rank submitted structural models of a protein through an explicit use of evolutionary information, a criterion differing from structural or energetic considerations commonly used in other assessment programs. The idea behind BeEP (Best Evolutionary Pattern) is to benefit from the substitution pattern derived from structural constraints present in a set of homologous proteins adopting a given protein conformation. The BeEP method uses a model of protein evolution that takes into account the structure of a protein to build site-specific substitution matrices. The suitability of these substitution matrices is assessed through maximum likelihood calculations from which position-specific and global scores can be derived. These scores estimate how well the structural constraints derived from each structural model are represented in a sequence alignment of homologous proteins. Our assessment on a subset of proteins from the Critical Assessment of techniques for protein Structure Prediction (CASP) experiment has shown that BeEP is capable of discriminating the models and selecting one or more native-like structures. Moreover, BeEP is not explicitly parameterized to find structural similarities between models and given targets, potentially helping to explore the conformational ensemble of the native state.
Figures


References
-
- Cozzetto D, Kryshtafovych A, Ceriani M, Tramontano A. Assessment of predictions in the model quality assessment category. Proteins. 2007;69(Suppl. 8):175–183. - PubMed
-
- Kryshtafovych A, Venclovas C, Fidelis K, Moult J. Progress over the first decade of CASP experiments. Proteins. 2005;61(Suppl. 7):225–236. - PubMed
-
- Moult J, Fidelis K, Kryshtafovych A, Rost B, Tramontano A. Critical assessment of methods of protein structure prediction - Round VIII. Proteins. 2009;77(Suppl. 9):1–4. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
