

Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis
- PMID: 23731542
- PMCID: PMC3675260
- DOI: 10.1016/j.ajhg.2013.04.024
Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis
Abstract
Infantile myofibromatosis (IM) is a disorder of mesenchymal proliferation characterized by the development of nonmetastasizing tumors in the skin, muscle, bone, and viscera. Occurrence within families across multiple generations is suggestive of an autosomal-dominant (AD) inheritance pattern, but autosomal-recessive (AR) modes of inheritance have also been proposed. We performed whole-exome sequencing (WES) in members of nine unrelated families clinically diagnosed with AD IM to identify the genetic origin of the disorder. In eight of the families, we identified one of two disease-causing mutations, c.1978C>A (p.Pro660Thr) and c.1681C>T (p.Arg561Cys), in PDGFRB. Intriguingly, one family did not have either of these PDGFRB mutations but all affected individuals had a c.4556T>C (p.Leu1519Pro) mutation in NOTCH3. Our studies suggest that mutations in PDGFRB are a cause of IM and highlight NOTCH3 as a candidate gene. Further studies of the crosstalk between PDGFRB and NOTCH pathways may offer new opportunities to identify mutations in other genes that result in IM and is a necessary first step toward understanding the mechanisms of both tumor growth and regression and its targeted treatment.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
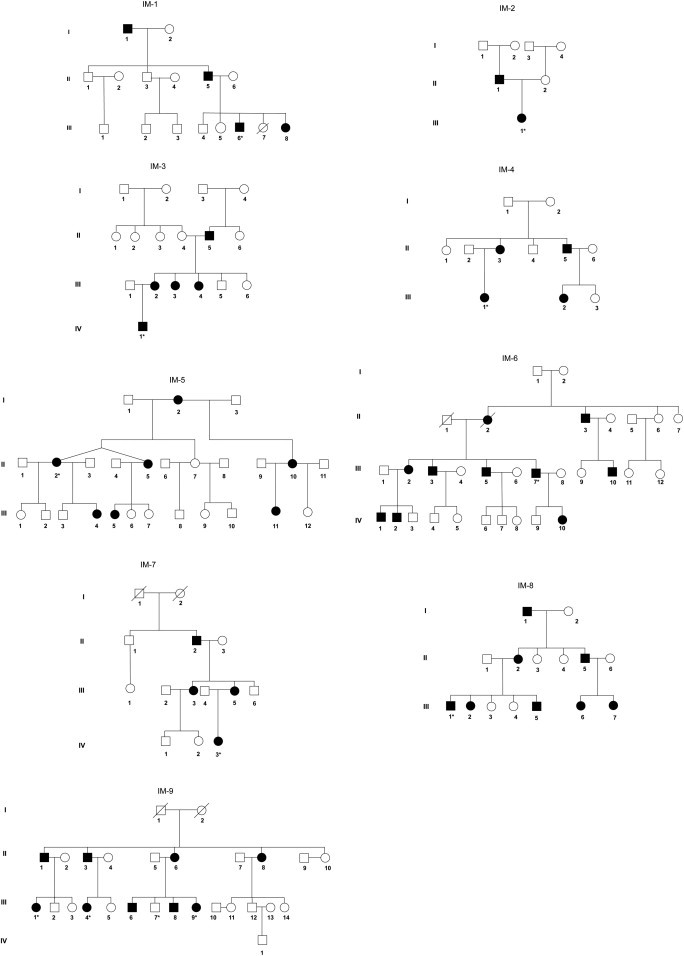
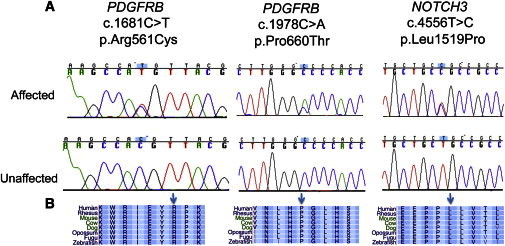
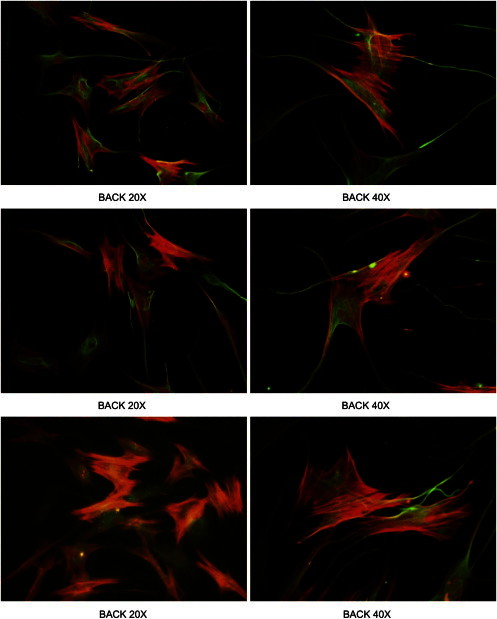
Comment in
-
Mutations in PDGFRB and NOTCH3 are the first genetic causes identified for autosomal dominant infantile myofibromatosis.Clin Genet. 2013 Oct;84(4):340-1. doi: 10.1111/cge.12238. Epub 2013 Jul 31. Clin Genet. 2013. PMID: 23865785 No abstract available.
-
[Autosomal dominant (familial) infantile myofibromatosis: The causative role of mutations in PDGFRB and NOTCH3].Ann Dermatol Venereol. 2013 Dec;140(12):833-4. doi: 10.1016/j.annder.2013.08.004. Epub 2013 Sep 26. Ann Dermatol Venereol. 2013. PMID: 24315235 French. No abstract available.
References
-
- Williams J.O., Schrum D. Congenital fibrosarcoma; report of a case in a newborn infant. AMA Arch. Pathol. 1951;51:548–552. - PubMed
-
- Stout A.P. Juvenile fibromatoses. Cancer. 1954;7:953–978. - PubMed
-
- Kauffman S.L., Stout A.P. Congenital mesenchymal tumors. Cancer. 1965;18:460–476. - PubMed
-
- Enzinger F.M. Fibrous hamartoma of infancy. Cancer. 1965;18:241–248. - PubMed
-
- Chung E.B., Enzinger F.M. Infantile myofibromatosis. Cancer. 1981;48:1807–1818. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
