

Pathophysiology of the diabetic kidney
- PMID: 23733640
- PMCID: PMC6029262
- DOI: 10.1002/cphy.c100049
Pathophysiology of the diabetic kidney
Abstract
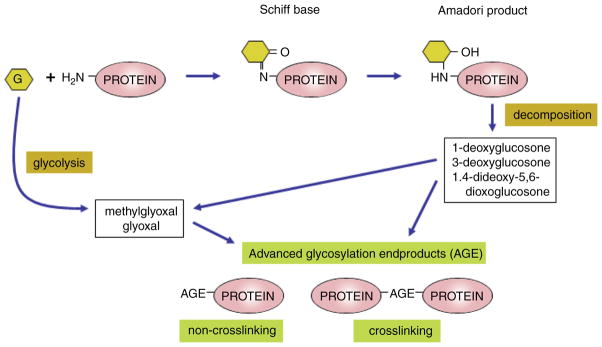
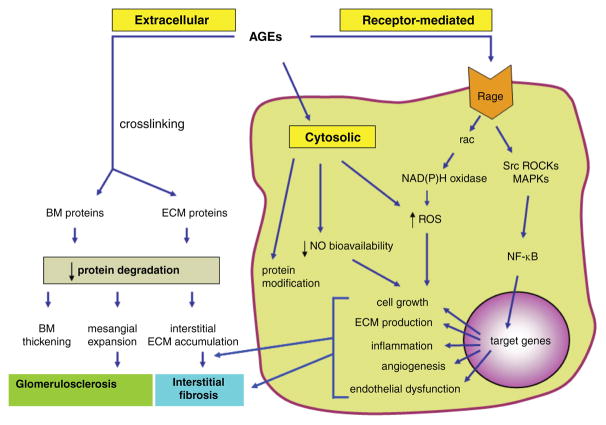
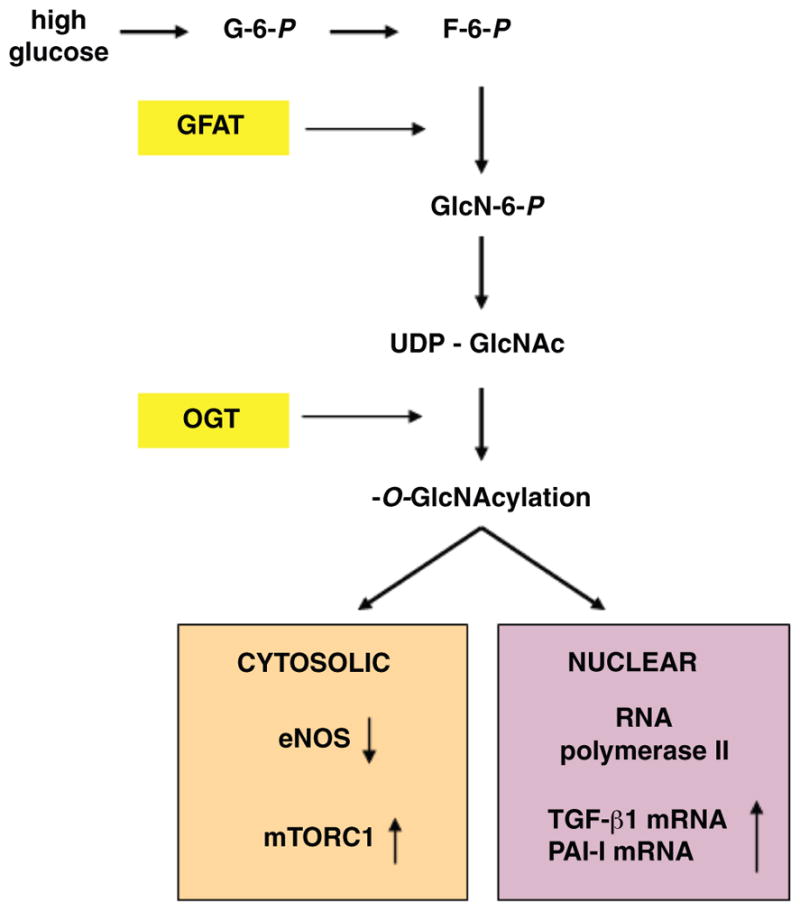
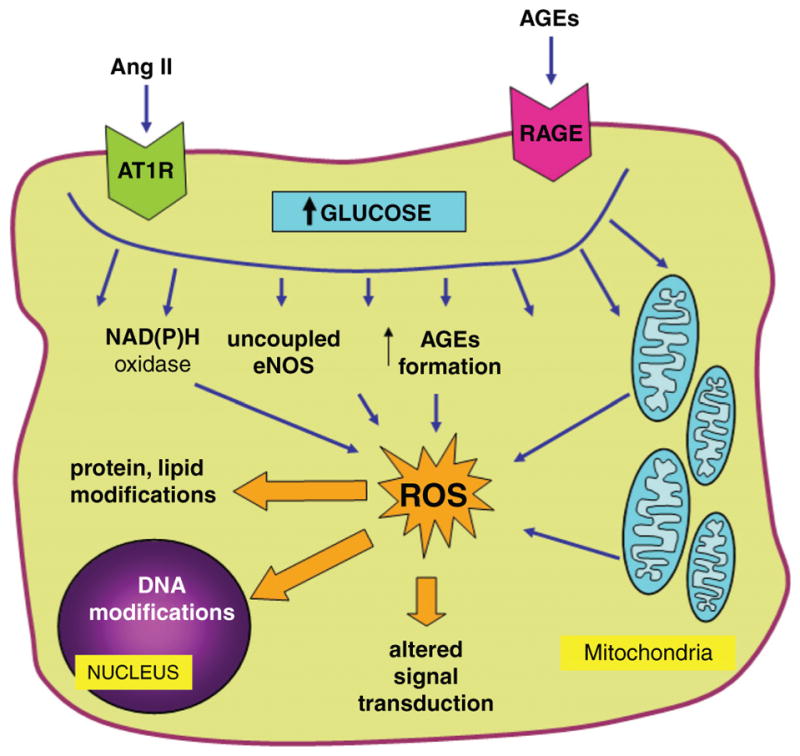
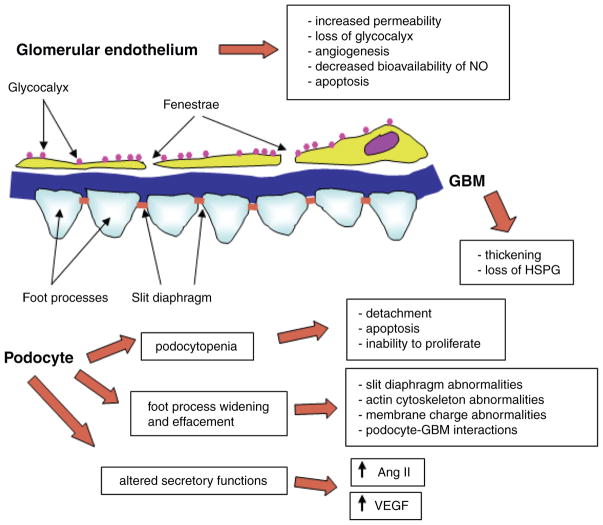
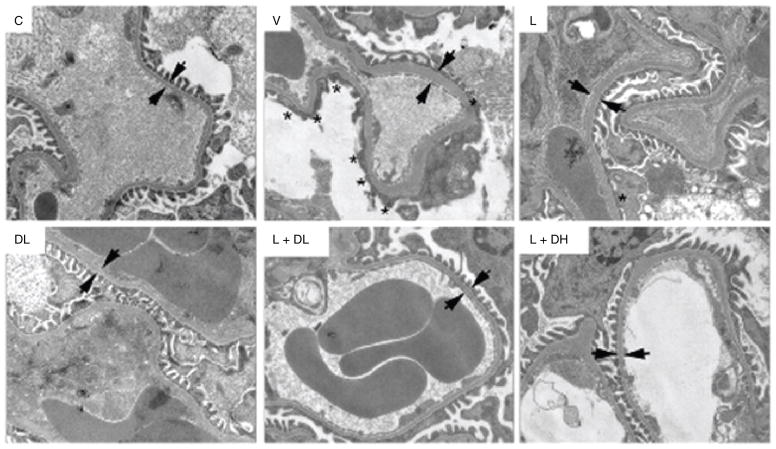
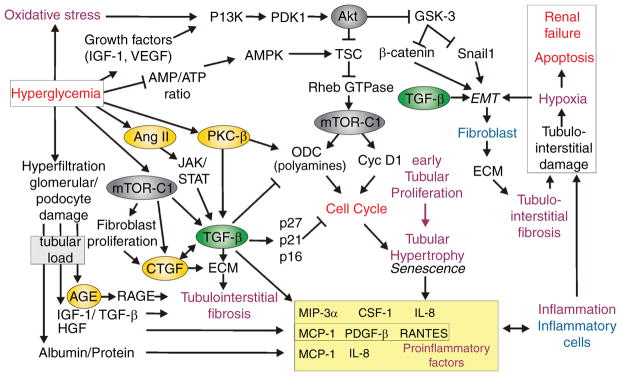
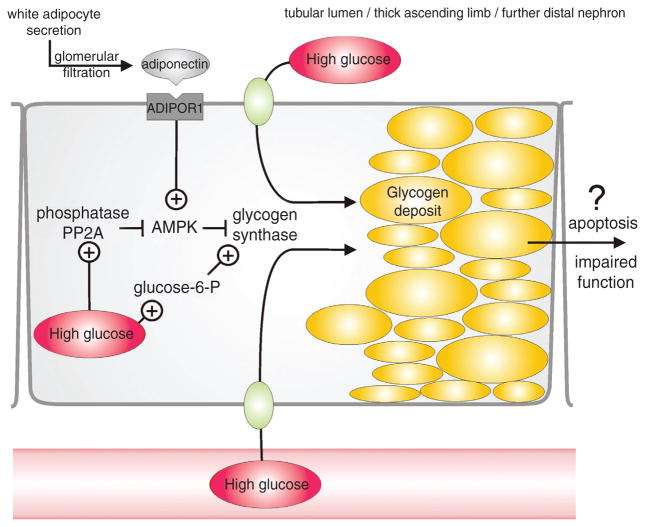
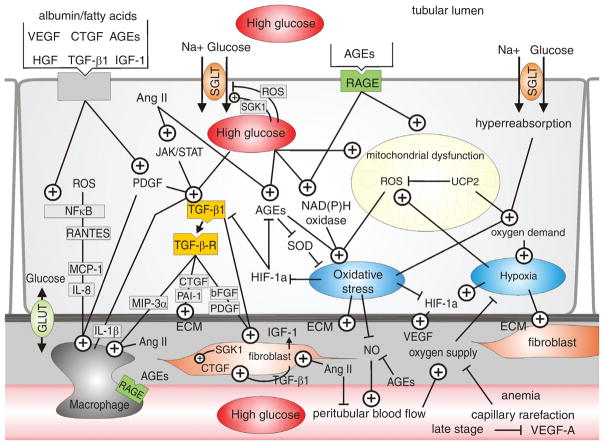
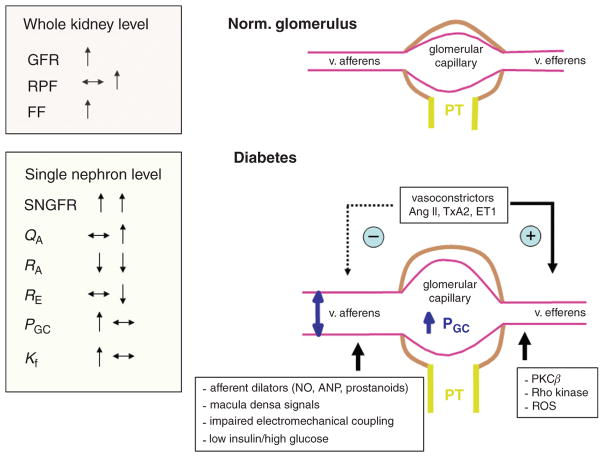
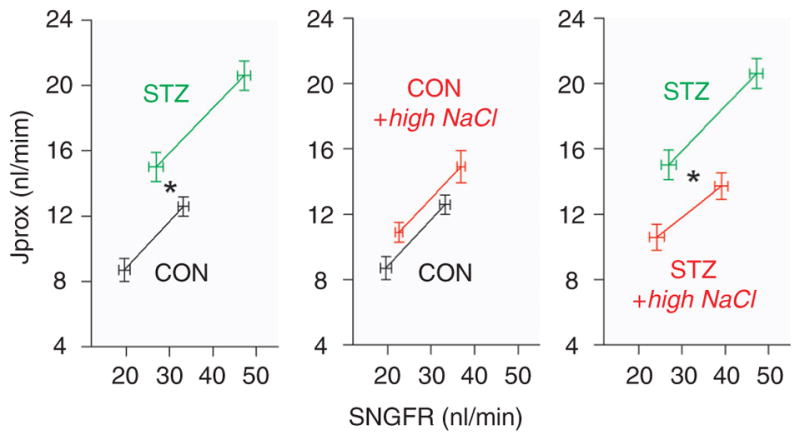
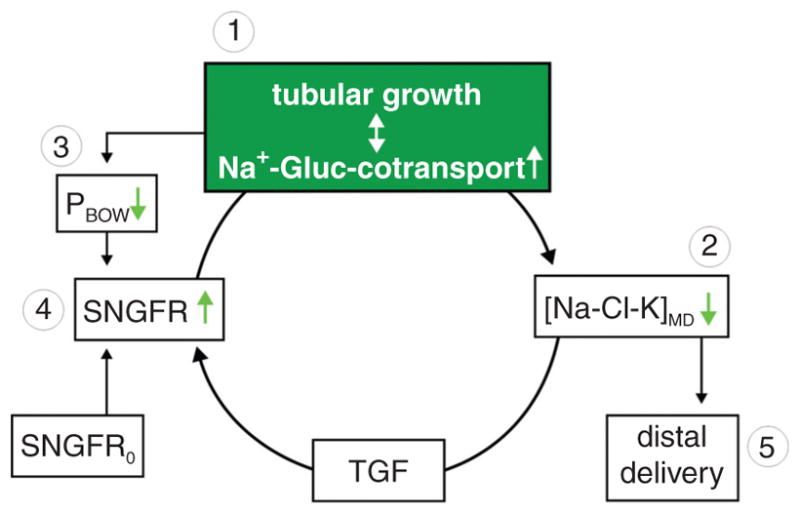
Diabetes mellitus contributes greatly to morbidity, mortality, and overall health care costs. In major part, these outcomes derive from the high incidence of progressive kidney dysfunction in patients with diabetes making diabetic nephropathy a leading cause of end-stage renal disease. A better understanding of the molecular mechanism involved and of the early dysfunctions observed in the diabetic kidney may permit the development of new strategies to prevent diabetic nephropathy. Here we review the pathophysiological changes that occur in the kidney in response to hyperglycemia, including the cellular responses to high glucose and the responses in vascular, glomerular, podocyte, and tubular function. The molecular basis, characteristics, and consequences of the unique growth phenotypes observed in the diabetic kidney, including glomerular structures and tubular segments, are outlined. We delineate mechanisms of early diabetic glomerular hyperfiltration including primary vascular events as well as the primary role of tubular growth, hyperreabsorption, and tubuloglomerular communication as part of a "tubulocentric" concept of early diabetic kidney function. The latter also explains the "salt paradox" of the early diabetic kidney, that is, a unique and inverse relationship between glomerular filtration rate and dietary salt intake. The mechanisms and consequences of the intrarenal activation of the renin-angiotensin system and of diabetes-induced tubular glycogen accumulation are discussed. Moreover, we aim to link the changes that occur early in the diabetic kidney including the growth phenotype, oxidative stress, hypoxia, and formation of advanced glycation end products to mechanisms involved in progressive kidney disease.
© 2011 American Physiological Society.
Figures
References
-
- Aaltonen P, Luimula P, Astrom E, Palmen T, Gronholm T, Palojoki E, Jaakkola I, Ahola H, Tikkanen I, Holthofer H. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab Invest. 2001;81:1185–1190. - PubMed
-
- Abbate M, Remuzzi G. Proteinuria as a mediator of tubulointerstitial injury. Kidney Blood Press Res. 1999;22:37–46. - PubMed
-
- Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol. 1998;9:1213–1224. - PubMed
-
- Abdel-Wahab N, Weston BS, Roberts T, Mason RM. Connective tissue growth factor and regulation of the mesangial cell cycle: Role in cellular hypertrophy. J Am Soc Nephrol. 2002;13:2437–2445. - PubMed
-
- Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med. 2005;39:1–25. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
