

Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections
- PMID: 23734239
- PMCID: PMC3666980
- DOI: 10.1371/journal.pone.0065226
Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections
Abstract
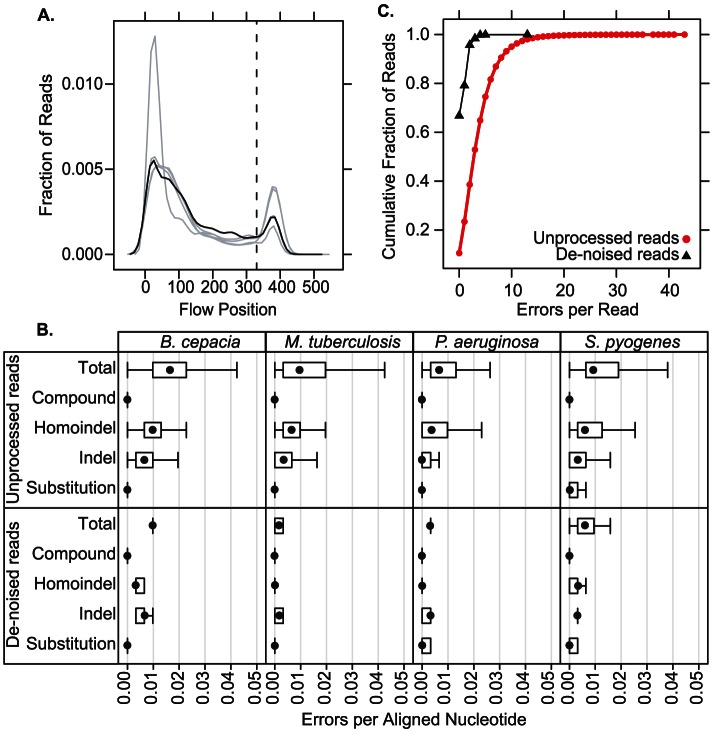
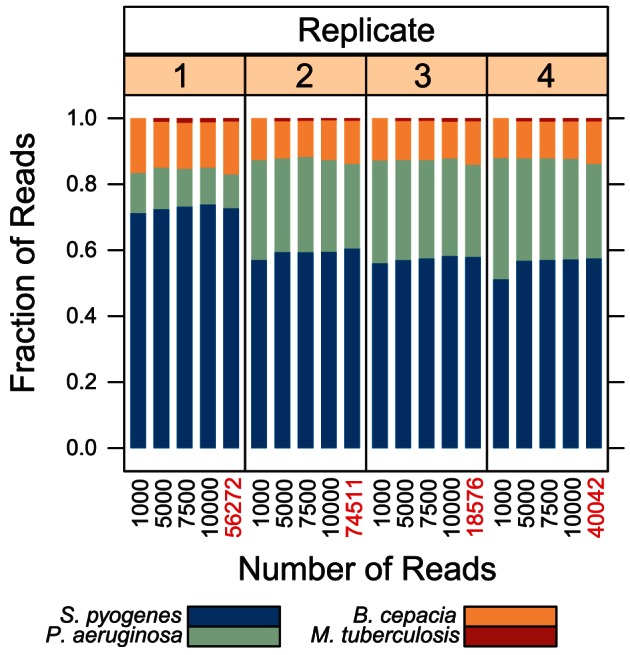
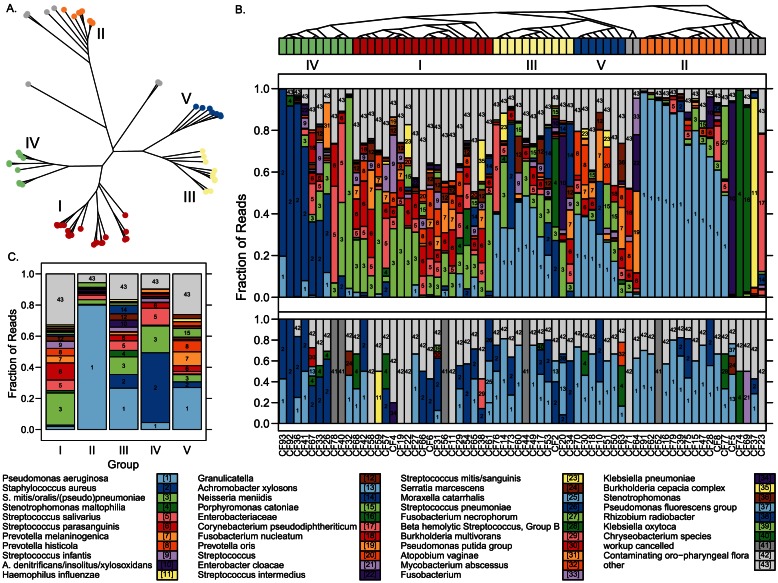
Classifying individual bacterial species comprising complex, polymicrobial patient specimens remains a challenge for culture-based and molecular microbiology techniques in common clinical use. We therefore adapted practices from metagenomics research to rapidly catalog the bacterial composition of clinical specimens directly from patients, without need for prior culture. We have combined a semiconductor deep sequencing protocol that produces reads spanning 16S ribosomal RNA gene variable regions 1 and 2 (∼360 bp) with a de-noising pipeline that significantly improves the fraction of error-free sequences. The resulting sequences can be used to perform accurate genus- or species-level taxonomic assignment. We explore the microbial composition of challenging, heterogeneous clinical specimens by deep sequencing, culture-based strain typing, and Sanger sequencing of bulk PCR product. We report that deep sequencing can catalog bacterial species in mixed specimens from which usable data cannot be obtained by conventional clinical methods. Deep sequencing a collection of sputum samples from cystic fibrosis (CF) patients reveals well-described CF pathogens in specimens where they were not detected by standard clinical culture methods, especially for low-prevalence or fastidious bacteria. We also found that sputa submitted for CF diagnostic workup can be divided into a limited number of groups based on the phylogenetic composition of the airway microbiota, suggesting that metagenomic profiling may prove useful as a clinical diagnostic strategy in the future. The described method is sufficiently rapid (theoretically compatible with same-day turnaround times) and inexpensive for routine clinical use.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
