

Joint genotype inference with germline and somatic mutations
- PMID: 23734724
- PMCID: PMC3622648
- DOI: 10.1186/1471-2105-14-S5-S3
Joint genotype inference with germline and somatic mutations
Abstract
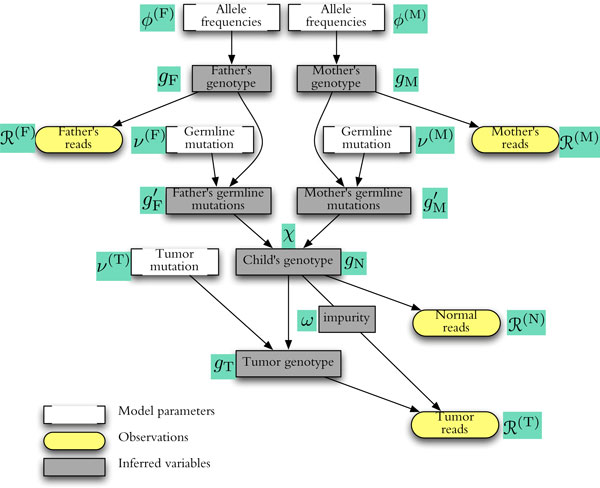
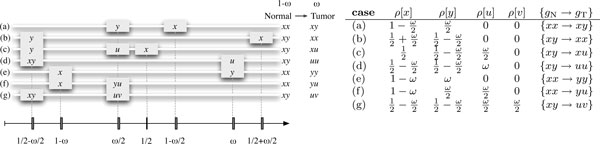
The joint sequencing of related genomes has become an important means to discover rare variants. Normal-tumor genome pairs are routinely sequenced together to find somatic mutations and their associations with different cancers. Parental and sibling genomes reveal de novo germline mutations and inheritance patterns related to Mendelian diseases.Acute lymphoblastic leukemia (ALL) is the most common paediatric cancer and the leading cause of cancer-related death among children. With the aim of uncovering the full spectrum of germline and somatic genetic alterations in childhood ALL genomes, we conducted whole-exome re-sequencing on a unique cohort of over 120 exomes of childhood ALL quartets, each comprising a patient's tumor and matched-normal material, and DNA from both parents. We developed a general probabilistic model for such quartet sequencing reads mapped to the reference human genome. The model is used to infer joint genotypes at homologous loci across a normal-tumor genome pair and two parental genomes.We describe the algorithms and data structures for genotype inference, model parameter training. We implemented the methods in an open-source software package (QUADGT) that uses the standard file formats of the 1000 Genomes Project. Our method's utility is illustrated on quartets from the ALL cohort.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
