

The structure of the Mycobacterium smegmatis trehalose synthase reveals an unusual active site configuration and acarbose-binding mode
- PMID: 23735230
- PMCID: PMC3724413
- DOI: 10.1093/glycob/cwt044
The structure of the Mycobacterium smegmatis trehalose synthase reveals an unusual active site configuration and acarbose-binding mode
Abstract
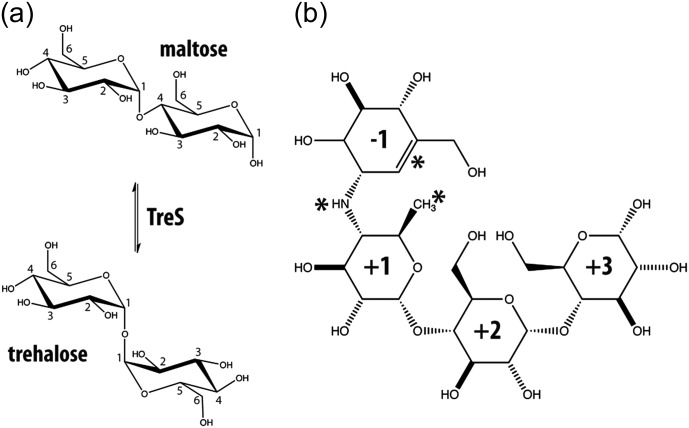
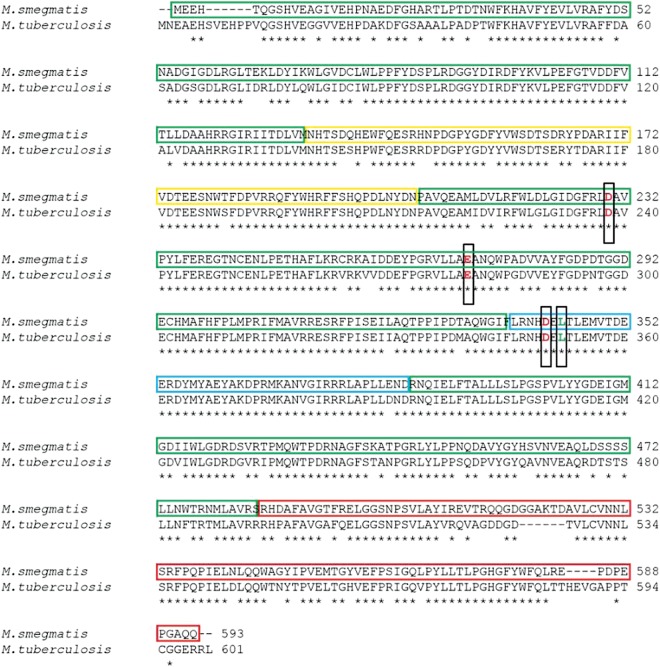
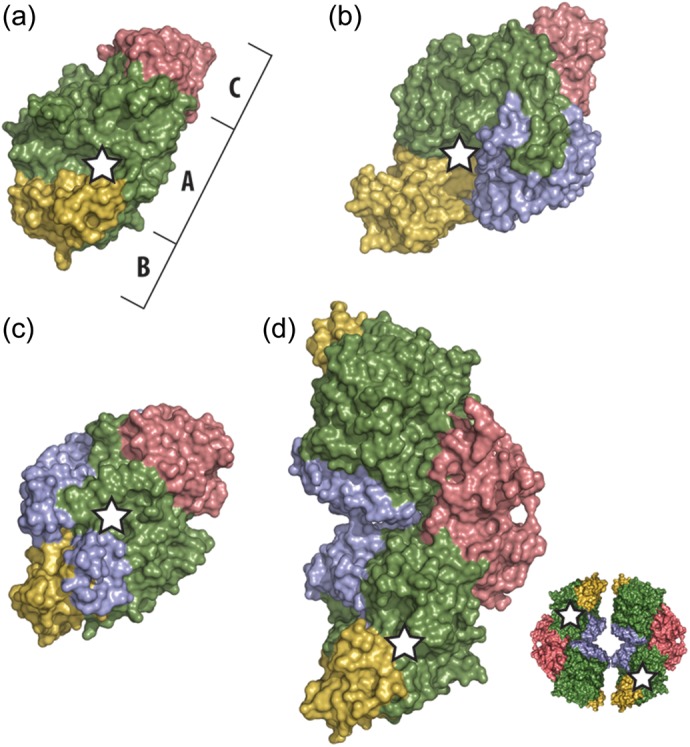
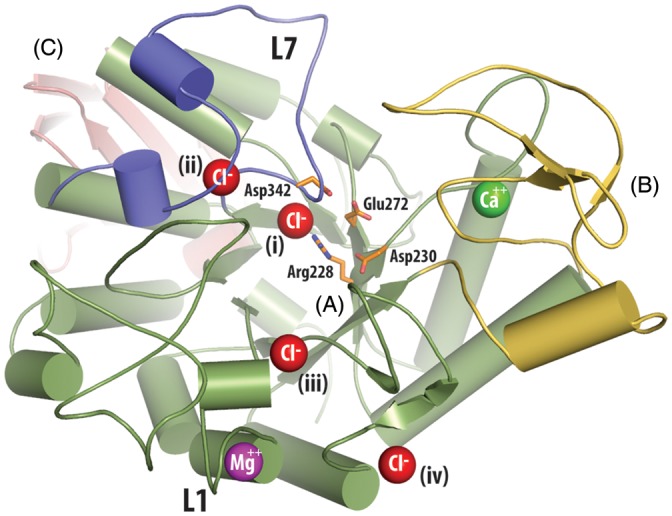
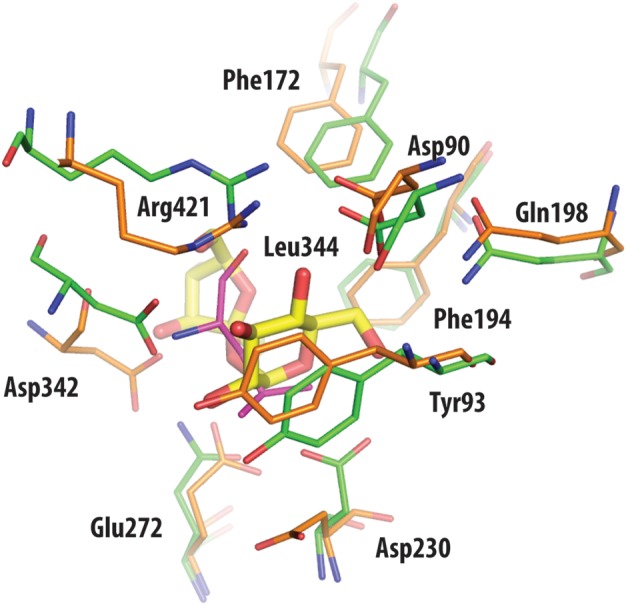
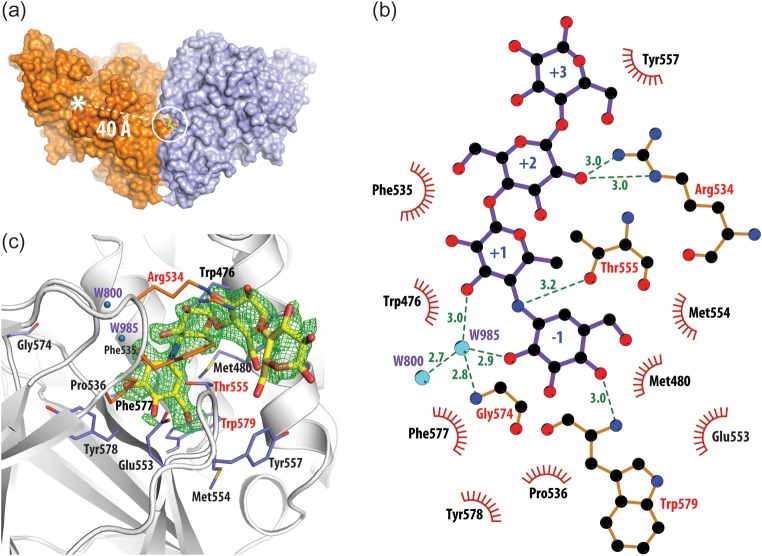
Trehalose synthase (TreS) catalyzes the reversible conversion of maltose into trehalose in mycobacteria as one of three biosynthetic pathways to this nonreducing disaccharide. Given the importance of trehalose to survival of mycobacteria, there has been considerable interest in understanding the enzymes involved in its production; indeed the structures of the key enzymes in the other two pathways have already been determined. Herein, we present the first structure of TreS from Mycobacterium smegmatis, thereby providing insights into the catalytic machinery involved in this intriguing intramolecular reaction. This structure, which is of interest both mechanistically and as a potential pharmaceutical target, reveals a narrow and enclosed active site pocket within which intramolecular substrate rearrangements can occur. We also present the structure of a complex of TreS with acarbose, revealing a hitherto unsuspected oligosaccharide-binding site within the C-terminal domain. This may well provide an anchor point for the association of TreS with glycogen, thereby enhancing its role in glycogen biosynthesis and degradation.
Keywords: GH13 glycoside hydrolase; drug design; enzyme inhibition; trehalose synthase; tuberculosis.
Figures

References
-
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi:10.1107/S0907444909052925. - DOI - PMC - PubMed
-
- Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, Corran PH, Furth AJ, Milman JD, Offord RE, et al. Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 angstrom resolution using amino acid sequence data. Nature. 1975;255:609–614. doi:10.1038/255609a0. - DOI - PubMed
-
- Barry CE, 3rd, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog Lipid Res. 1998;37:143–179. doi:10.1016/S0163-7827(98)00008-3. - DOI - PubMed
-
- Barry CE, III, Mdluli K. Drug sensitivity and environmental adaptation of mycobacterial cell wall components. Trends Microbiol. 1996;4:275–281. doi:10.1016/0966-842X(96)10031-7. - DOI - PubMed
-
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi:10.1042/BJ20040892. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
