

SOAP3-dp: fast, accurate and sensitive GPU-based short read aligner
- PMID: 23741504
- PMCID: PMC3669295
- DOI: 10.1371/journal.pone.0065632
SOAP3-dp: fast, accurate and sensitive GPU-based short read aligner
Erratum in
- PLoS One. 2013;8(8). doi:10.1371/annotation/823f3670-ed17-41ec-ba51-b50281651915
Abstract
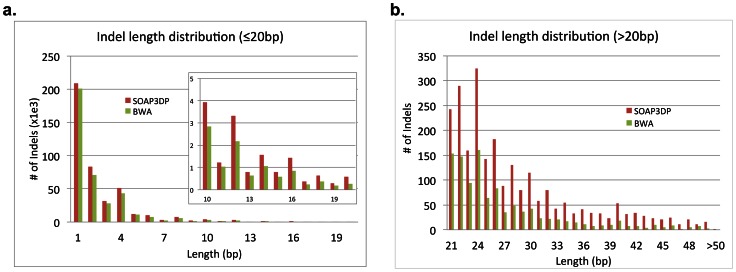
To tackle the exponentially increasing throughput of Next-Generation Sequencing (NGS), most of the existing short-read aligners can be configured to favor speed in trade of accuracy and sensitivity. SOAP3-dp, through leveraging the computational power of both CPU and GPU with optimized algorithms, delivers high speed and sensitivity simultaneously. Compared with widely adopted aligners including BWA, Bowtie2, SeqAlto, CUSHAW2, GEM and GPU-based aligners BarraCUDA and CUSHAW, SOAP3-dp was found to be two to tens of times faster, while maintaining the highest sensitivity and lowest false discovery rate (FDR) on Illumina reads with different lengths. Transcending its predecessor SOAP3, which does not allow gapped alignment, SOAP3-dp by default tolerates alignment similarity as low as 60%. Real data evaluation using human genome demonstrates SOAP3-dp's power to enable more authentic variants and longer Indels to be discovered. Fosmid sequencing shows a 9.1% FDR on newly discovered deletions. SOAP3-dp natively supports BAM file format and provides the same scoring scheme as BWA, which enables it to be integrated into existing analysis pipelines. SOAP3-dp has been deployed on Amazon-EC2, NIH-Biowulf and Tianhe-1A.
Conflict of interest statement
Figures
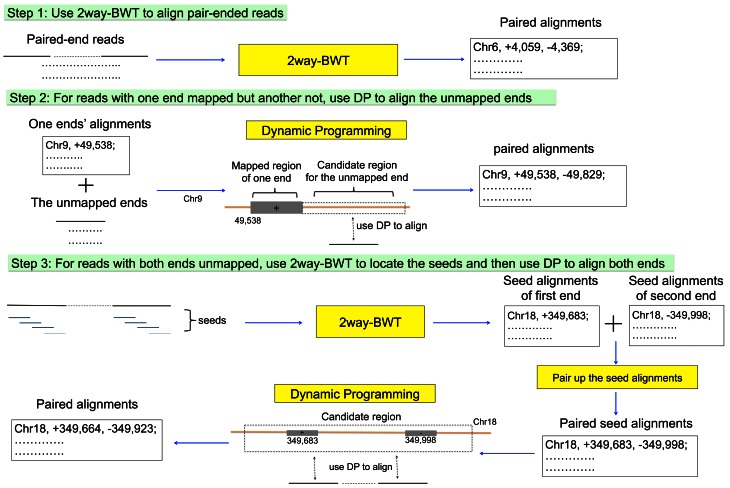
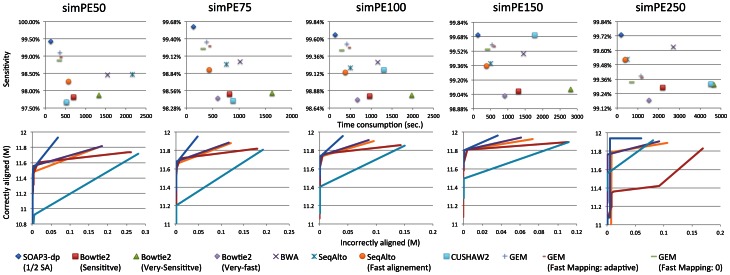
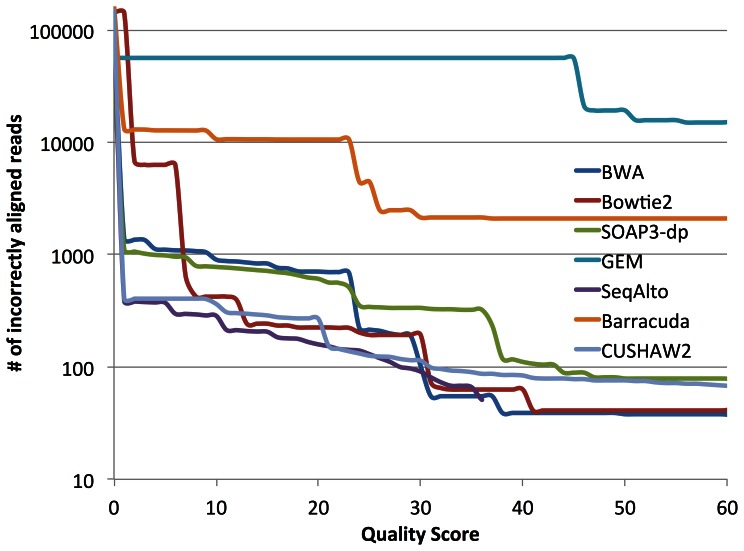
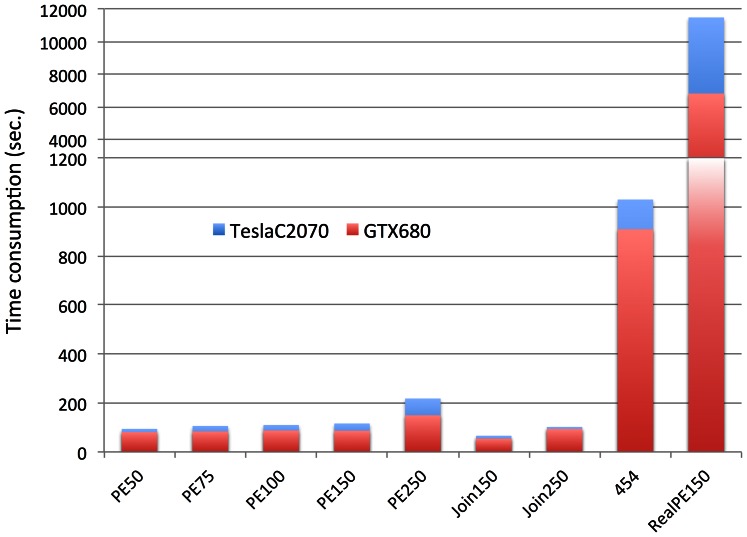
References
-
- Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
