

Long runs of homozygosity are enriched for deleterious variation
- PMID: 23746547
- PMCID: PMC3710769
- DOI: 10.1016/j.ajhg.2013.05.003
Long runs of homozygosity are enriched for deleterious variation
Abstract
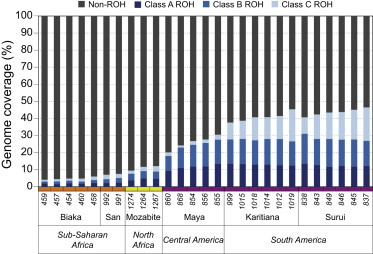
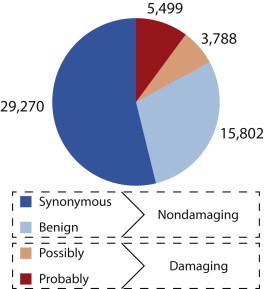
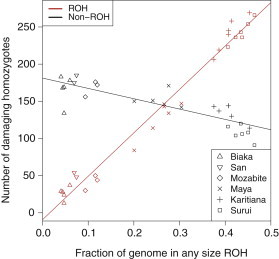
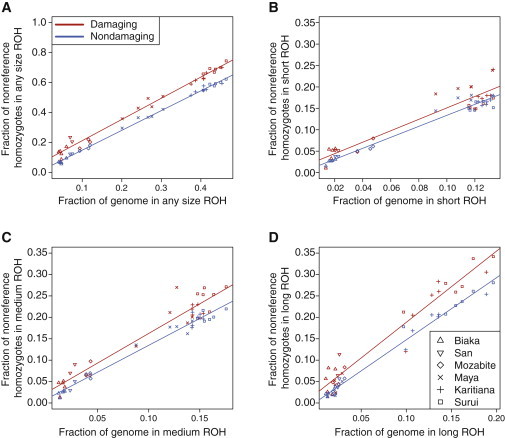
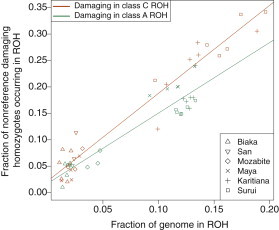
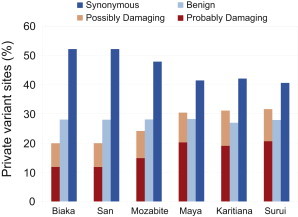
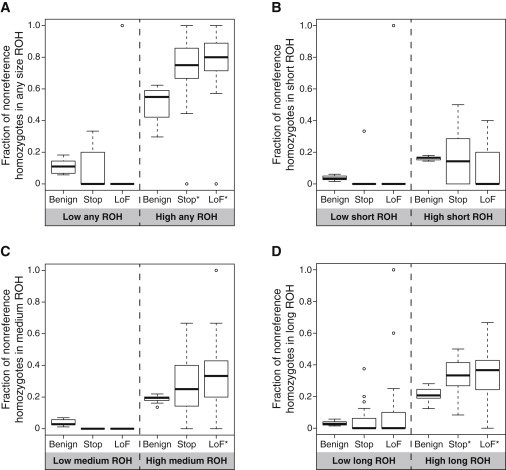
Exome sequencing offers the potential to study the population-genomic variables that underlie patterns of deleterious variation. Runs of homozygosity (ROH) are long stretches of consecutive homozygous genotypes probably reflecting segments shared identically by descent as the result of processes such as consanguinity, population size reduction, and natural selection. The relationship between ROH and patterns of predicted deleterious variation can provide insight into the way in which these processes contribute to the maintenance of deleterious variants. Here, we use exome sequencing to examine ROH in relation to the distribution of deleterious variation in 27 individuals of varying levels of apparent inbreeding from 6 human populations. A significantly greater fraction of all genome-wide predicted damaging homozygotes fall in ROH than would be expected from the corresponding fraction of nondamaging homozygotes in ROH (p < 0.001). This pattern is strongest for long ROH (p < 0.05). ROH, and especially long ROH, harbor disproportionately more deleterious homozygotes than would be expected on the basis of the total ROH coverage of the genome and the genomic distribution of nondamaging homozygotes. The results accord with a hypothesis that recent inbreeding, which generates long ROH, enables rare deleterious variants to exist in homozygous form. Thus, just as inbreeding can elevate the occurrence of rare recessive diseases that represent homozygotes for strongly deleterious mutations, inbreeding magnifies the occurrence of mildly deleterious variants as well.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Kondrashov A.S. Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 1995;175:583–594. - PubMed
-
- Charlesworth B., Charlesworth D. Some evolutionary consequences of deleterious mutations. Genetica. 1998;102-103:3–19. - PubMed
-
- Eyre-Walker A., Keightley P.D. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
