

Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy
- PMID: 23746549
- PMCID: PMC3710748
- DOI: 10.1016/j.ajhg.2013.05.004
Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy
Abstract
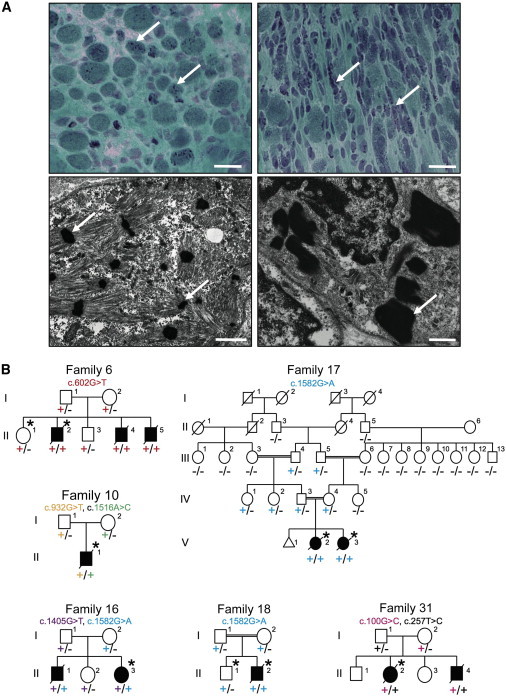
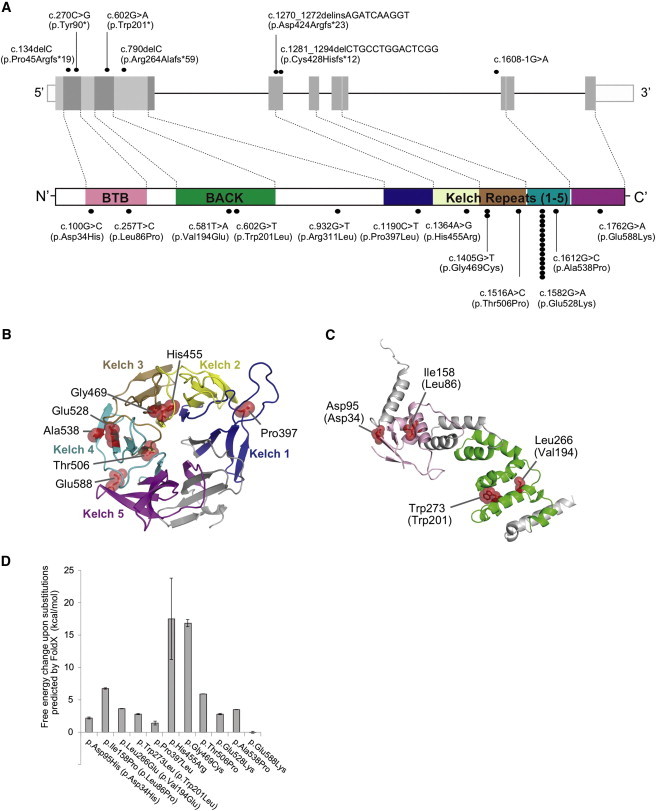
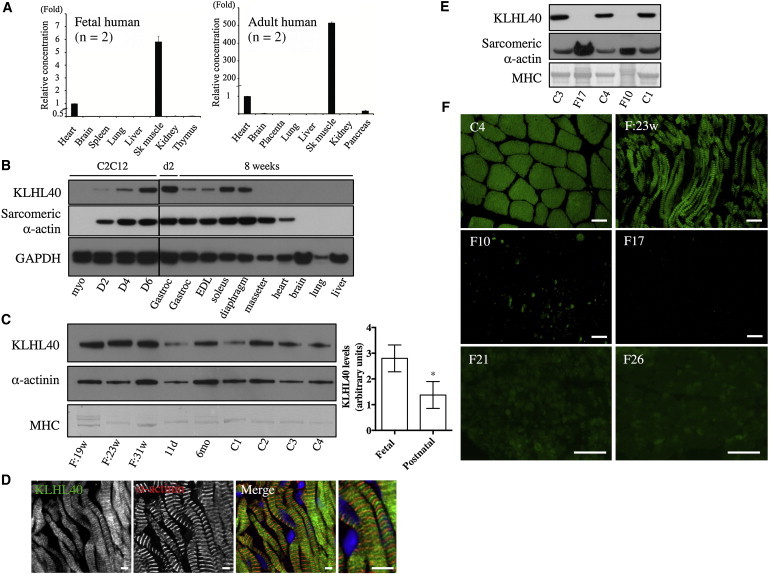
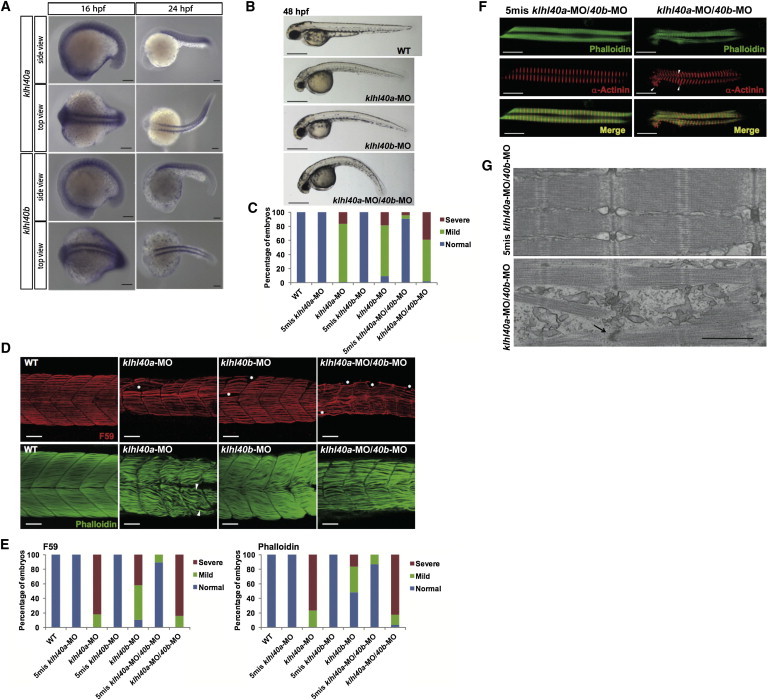
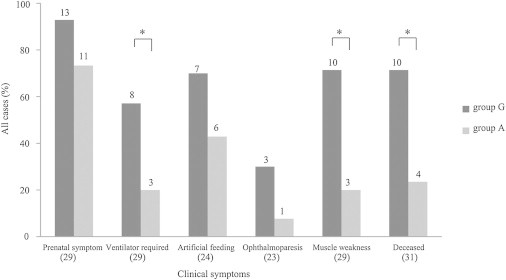
Nemaline myopathy (NEM) is a common congenital myopathy. At the very severe end of the NEM clinical spectrum are genetically unresolved cases of autosomal-recessive fetal akinesia sequence. We studied a multinational cohort of 143 severe-NEM-affected families lacking genetic diagnosis. We performed whole-exome sequencing of six families and targeted gene sequencing of additional families. We identified 19 mutations in KLHL40 (kelch-like family member 40) in 28 apparently unrelated NEM kindreds of various ethnicities. Accounting for up to 28% of the tested individuals in the Japanese cohort, KLHL40 mutations were found to be the most common cause of this severe form of NEM. Clinical features of affected individuals were severe and distinctive and included fetal akinesia or hypokinesia and contractures, fractures, respiratory failure, and swallowing difficulties at birth. Molecular modeling suggested that the missense substitutions would destabilize the protein. Protein studies showed that KLHL40 is a striated-muscle-specific protein that is absent in KLHL40-associated NEM skeletal muscle. In zebrafish, klhl40a and klhl40b expression is largely confined to the myotome and skeletal muscle, and knockdown of these isoforms results in disruption of muscle structure and loss of movement. We identified KLHL40 mutations as a frequent cause of severe autosomal-recessive NEM and showed that it plays a key role in muscle development and function. Screening of KLHL40 should be a priority in individuals who are affected by autosomal-recessive NEM and who present with prenatal symptoms and/or contractures and in all Japanese individuals with severe NEM.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Nowak K.J., Wattanasirichaigoon D., Goebel H.H., Wilce M., Pelin K., Donner K., Jacob R.L., Hübner C., Oexle K., Anderson J.R. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 1999;23:208–212. - PubMed
-
- Agrawal P.B., Greenleaf R.S., Tomczak K.K., Lehtokari V.L., Wallgren-Pettersson C., Wallefeld W., Laing N.G., Darras B.T., Maciver S.K., Dormitzer P.R., Beggs A.H. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 2007;80:162–167. - PMC - PubMed
-
- Lehtokari V.L., Pelin K., Sandbacka M., Ranta S., Donner K., Muntoni F., Sewry C., Angelini C., Bushby K., Van den Bergh P. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum. Mutat. 2006;27:946–956. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
