

Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia
- PMID: 23746551
- PMCID: PMC3710753
- DOI: 10.1016/j.ajhg.2013.05.006
Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia
Abstract
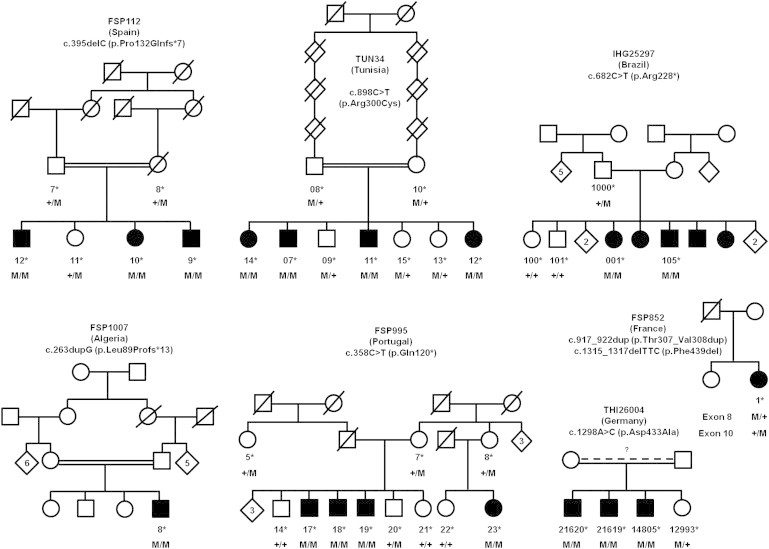
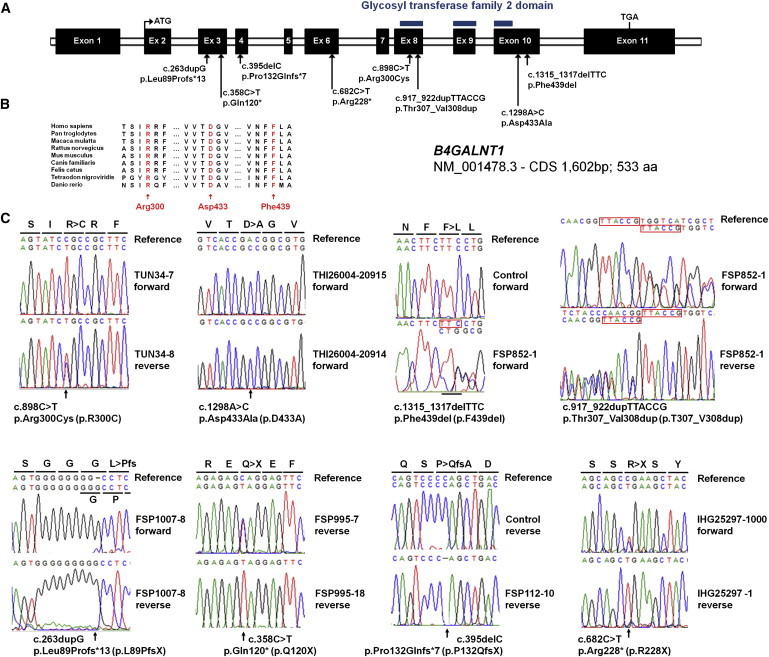
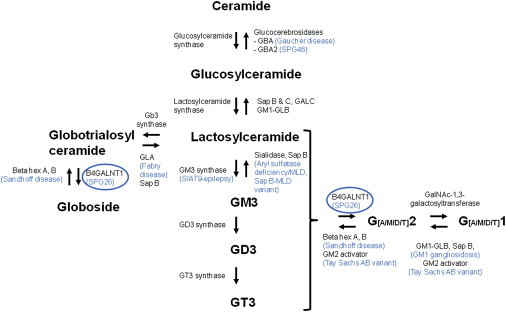
Hereditary spastic paraplegias (HSPs) form a heterogeneous group of neurological disorders. A whole-genome linkage mapping effort was made with three HSP-affected families from Spain, Portugal, and Tunisia and it allowed us to reduce the SPG26 locus interval from 34 to 9 Mb. Subsequently, a targeted capture was made to sequence the entire exome of affected individuals from these three families, as well as from two additional autosomal-recessive HSP-affected families of German and Brazilian origins. Five homozygous truncating (n = 3) and missense (n = 2) mutations were identified in B4GALNT1. After this finding, we analyzed the entire coding region of this gene in 65 additional cases, and three mutations were identified in two subjects. All mutated cases presented an early-onset spastic paraplegia, with frequent intellectual disability, cerebellar ataxia, and peripheral neuropathy as well as cortical atrophy and white matter hyperintensities on brain imaging. B4GALNT1 encodes β-1,4-N-acetyl-galactosaminyl transferase 1 (B4GALNT1), involved in ganglioside biosynthesis. These findings confirm the increasing interest of lipid metabolism in HSPs. Interestingly, although the catabolism of gangliosides is implicated in a variety of neurological diseases, SPG26 is only the second human disease involving defects of their biosynthesis.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Stevanin G., Ruberg M., Brice A. Recent advances in the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2008;8:198–210. - PubMed
-
- Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. - PubMed
-
- Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. - PubMed
-
- Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
