

Genome-wide inference of natural selection on human transcription factor binding sites
- PMID: 23749186
- PMCID: PMC3932982
- DOI: 10.1038/ng.2658
Genome-wide inference of natural selection on human transcription factor binding sites
Abstract
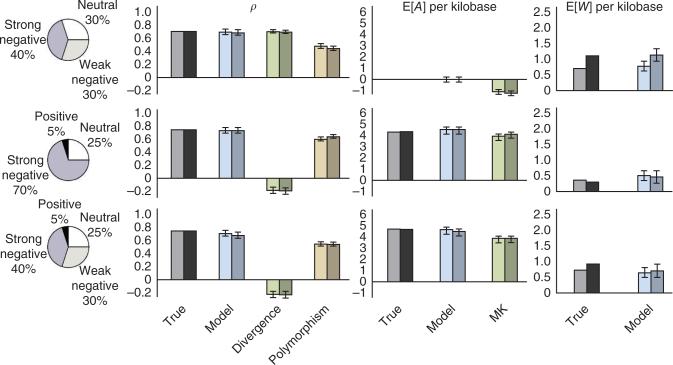
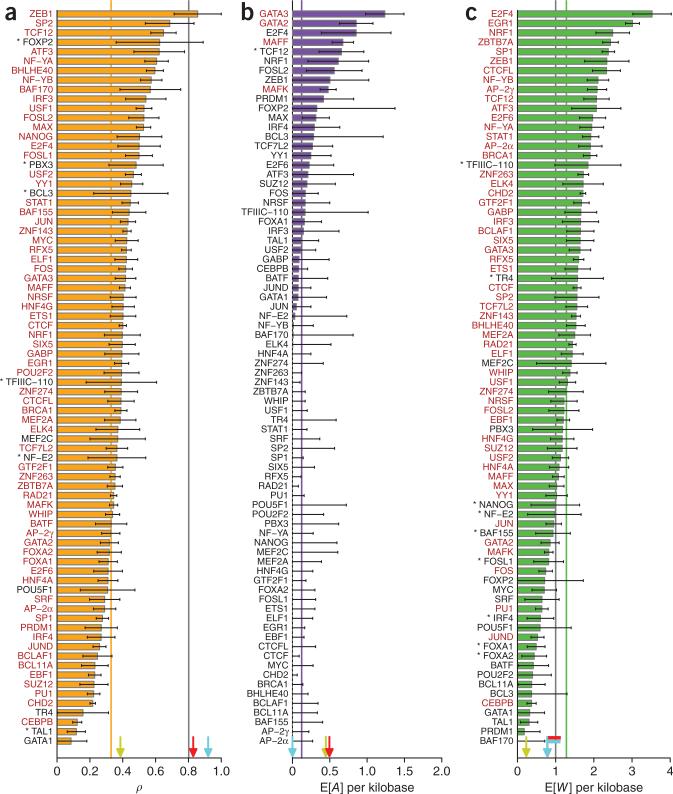
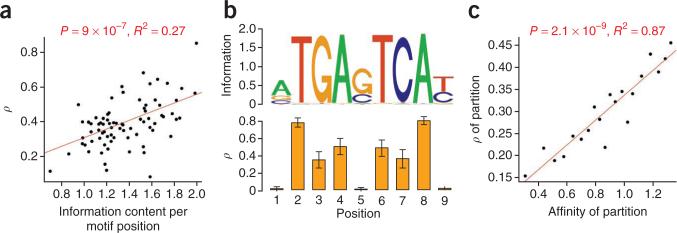
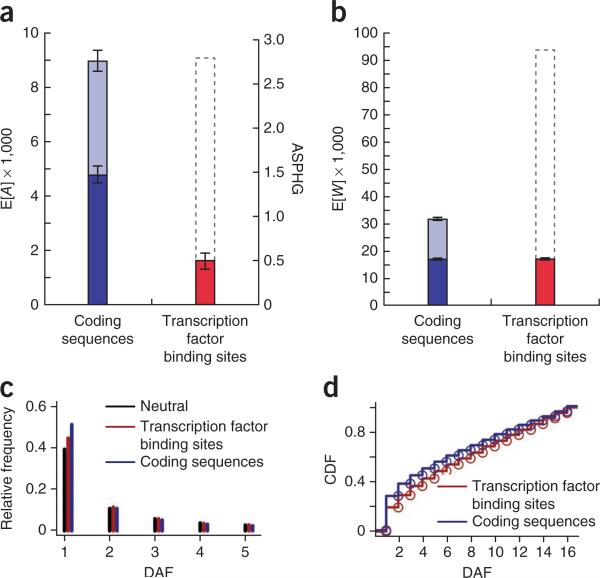
For decades, it has been hypothesized that gene regulation has had a central role in human evolution, yet much remains unknown about the genome-wide impact of regulatory mutations. Here we use whole-genome sequences and genome-wide chromatin immunoprecipitation and sequencing data to demonstrate that natural selection has profoundly influenced human transcription factor binding sites since the divergence of humans from chimpanzees 4-6 million years ago. Our analysis uses a new probabilistic method, called INSIGHT, for measuring the influence of selection on collections of short, interspersed noncoding elements. We find that, on average, transcription factor binding sites have experienced somewhat weaker selection than protein-coding genes. However, the binding sites of several transcription factors show clear evidence of adaptation. Several measures of selection are strongly correlated with predicted binding affinity. Overall, regulatory elements seem to contribute substantially to both adaptive substitutions and deleterious polymorphisms with key implications for human evolution and disease.
Figures
Comment in
-
Genome evolution: Adaptive human regulatory variation.Nat Rev Genet. 2013 Aug;14(8):520. doi: 10.1038/nrg3537. Epub 2013 Jun 25. Nat Rev Genet. 2013. PMID: 23797852 No abstract available.
References
-
- Ohno S. An argument for the genetic simplicity of man and other mammals. J. Hum. Evol. 1972;1:651–662.
-
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. - PubMed
-
- Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165:349–357. - PubMed
-
- Stern DL. Evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
