

The Growing Importance of CNVs: New Insights for Detection and Clinical Interpretation
- PMID: 23750167
- PMCID: PMC3667386
- DOI: 10.3389/fgene.2013.00092
The Growing Importance of CNVs: New Insights for Detection and Clinical Interpretation
Abstract
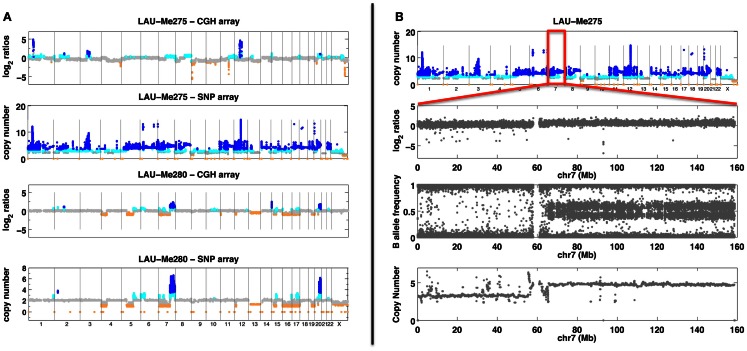
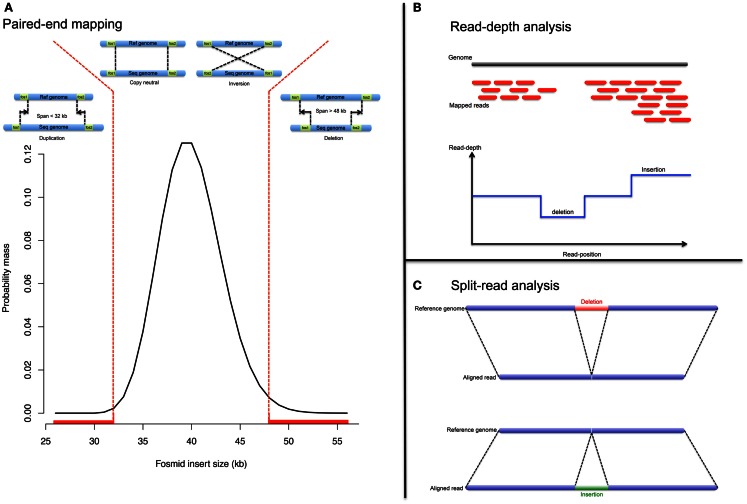
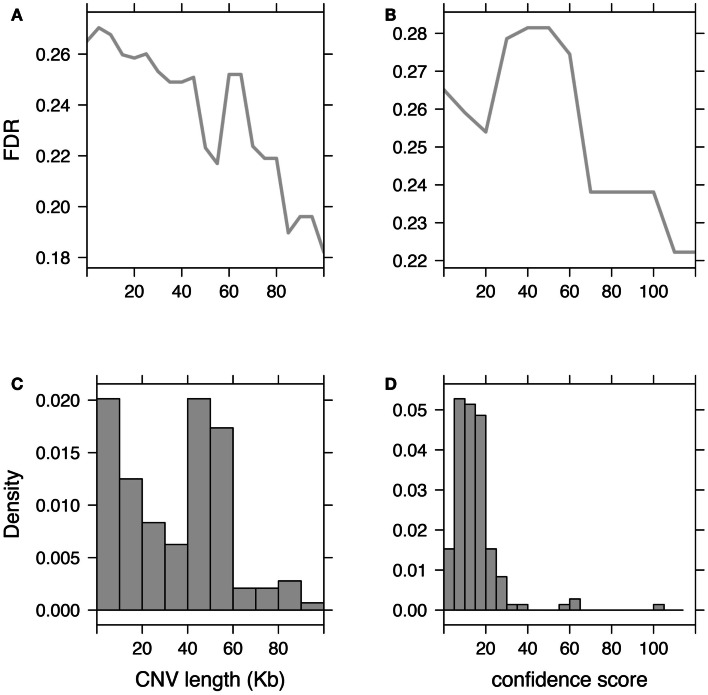
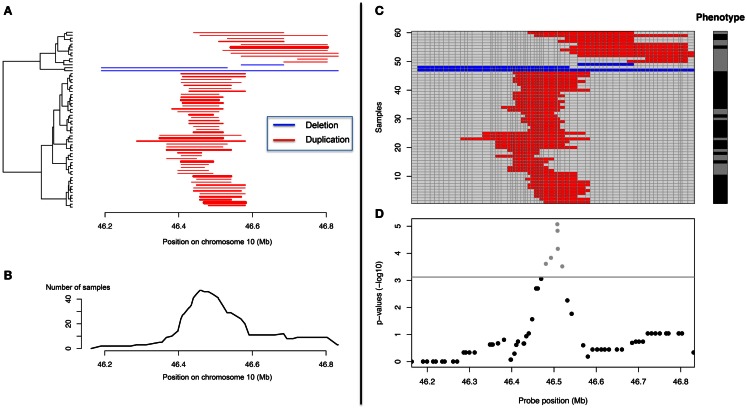
Differences between genomes can be due to single nucleotide variants, translocations, inversions, and copy number variants (CNVs, gain or loss of DNA). The latter can range from sub-microscopic events to complete chromosomal aneuploidies. Small CNVs are often benign but those larger than 500 kb are strongly associated with morbid consequences such as developmental disorders and cancer. Detecting CNVs within and between populations is essential to better understand the plasticity of our genome and to elucidate its possible contribution to disease. Hence there is a need for better-tailored and more robust tools for the detection and genome-wide analyses of CNVs. While a link between a given CNV and a disease may have often been established, the relative CNV contribution to disease progression and impact on drug response is not necessarily understood. In this review we discuss the progress, challenges, and limitations that occur at different stages of CNV analysis from the detection (using DNA microarrays and next-generation sequencing) and identification of recurrent CNVs to the association with phenotypes. We emphasize the importance of germline CNVs and propose strategies to aid clinicians to better interpret structural variations and assess their clinical implications.
Keywords: bioinformatics; complex disease; copy number variation; genome-wide association studies; genomics; personalized medicine; sequencing.
Figures
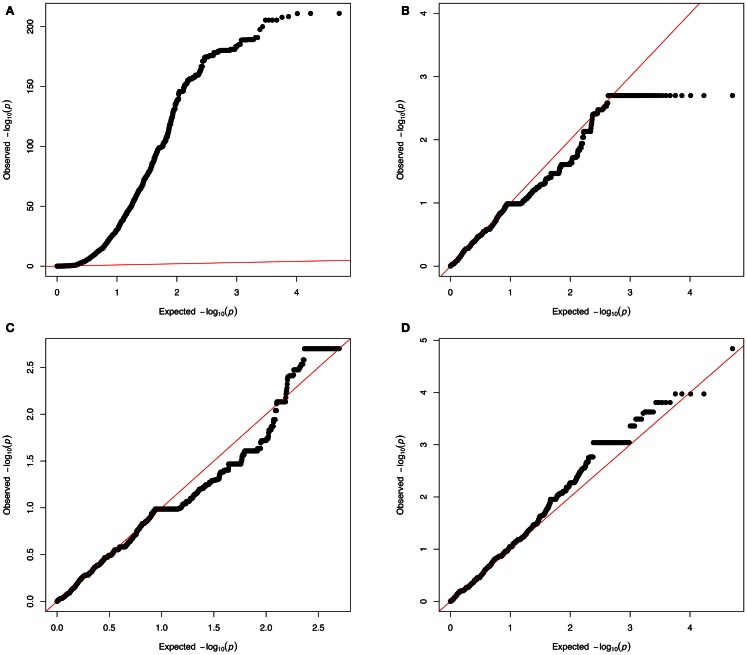
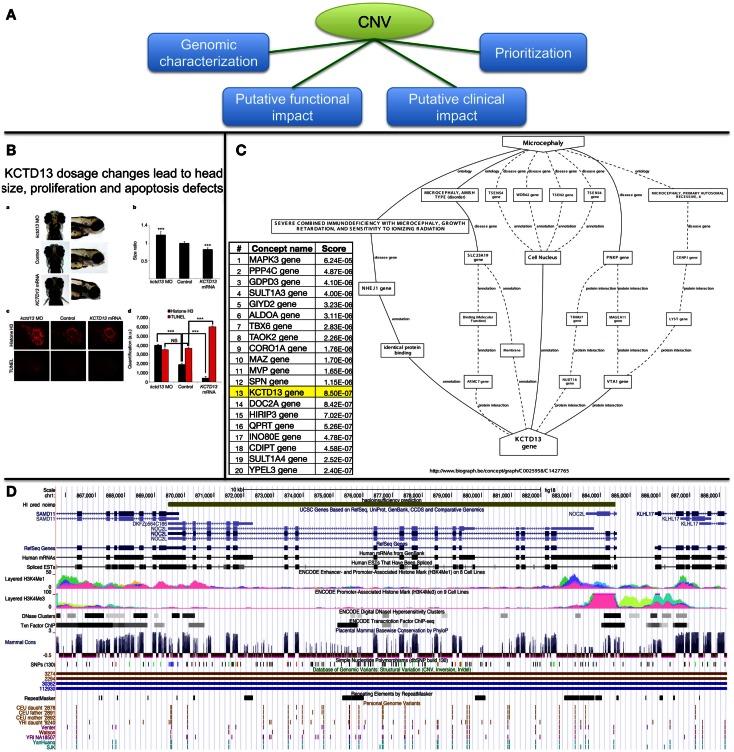
References
-
- Attiyeh E. F., Diskin S. J., Attiyeh M. A., Mossé Y. P., Hou C., Jackson E. M., et al. (2009). Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 19, 276–283 10.1101/gr.075671.107 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources