

DNA methylation data analysis and its application to cancer research
- PMID: 23750645
- PMCID: PMC4525706
- DOI: 10.2217/epi.13.26
DNA methylation data analysis and its application to cancer research
Abstract
With the rapid development of genome-wide high-throughput technologies, including expression arrays, SNP arrays and next-generation sequencing platforms, enormous amounts of molecular data have been generated and deposited in the public domain. The application of computational approaches is required to yield biological insights from this enormous, ever-growing resource. A particularly interesting subset of these resources is related to epigenetic regulation, with DNA methylation being the most abundant data type. In this paper, we will focus on the analysis of DNA methylation data and its application to cancer studies. We first briefly review the molecular techniques that generate such data, much of which has been obtained with the use of the most recent version of Infinium HumanMethylation450 BeadChip(®) technology (Illumina, CA, USA). We describe the coverage of the methylome by this technique. Several examples of data mining are provided. However, it should be understood that reliance on a single aspect of epigenetics has its limitations. In the not too distant future, these defects may be rectified, providing scientists with previously unavailable opportunities to explore in detail the role of epigenetics in cancer and other disease states.
Conflict of interest statement
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Figures
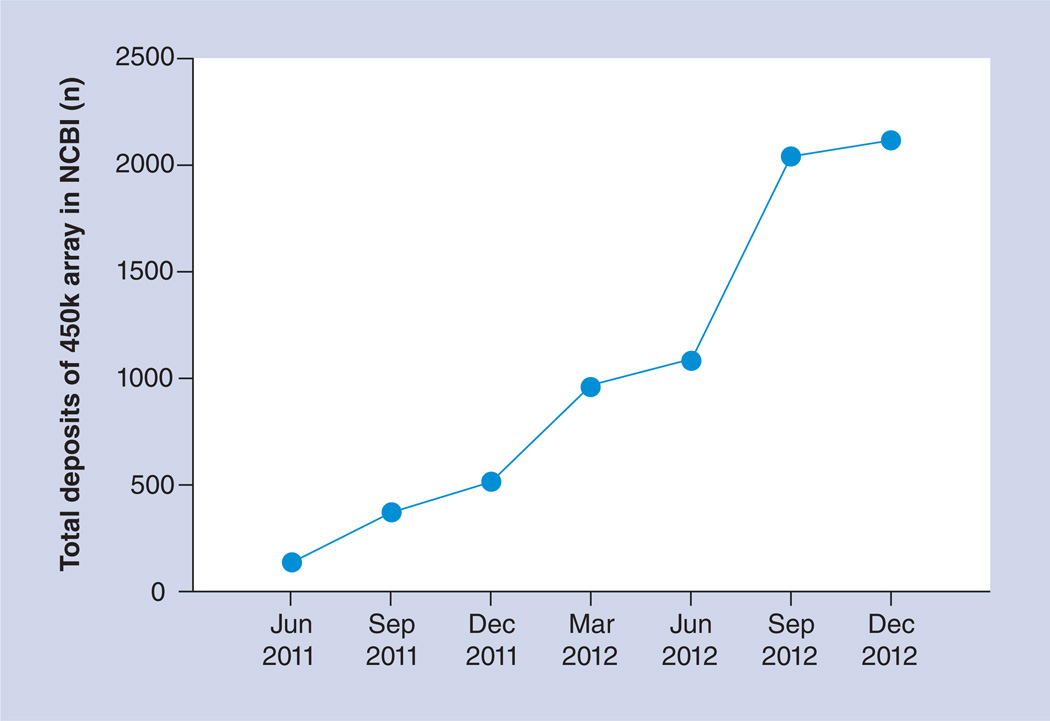
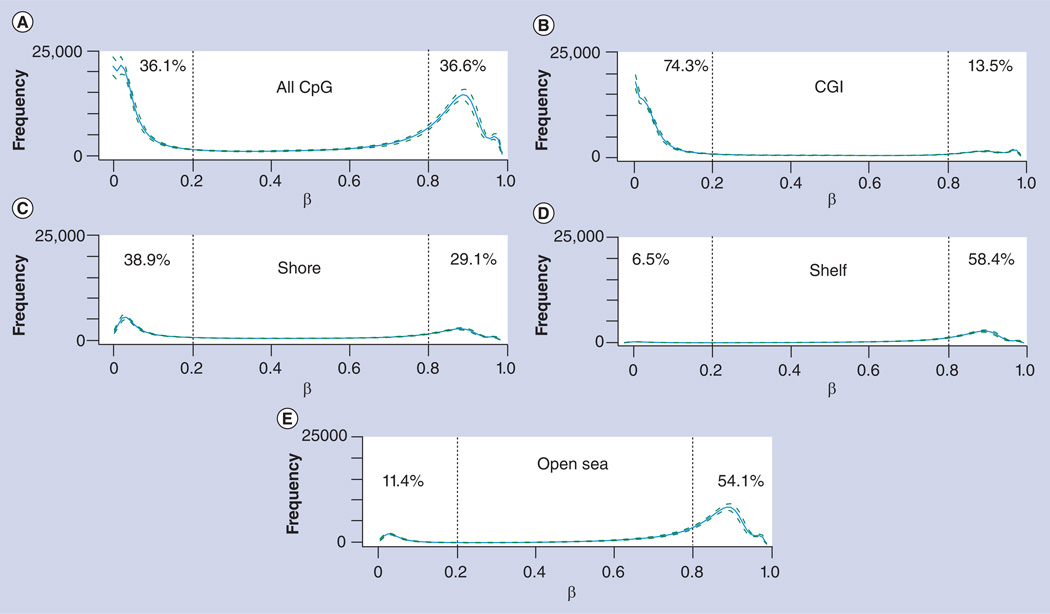
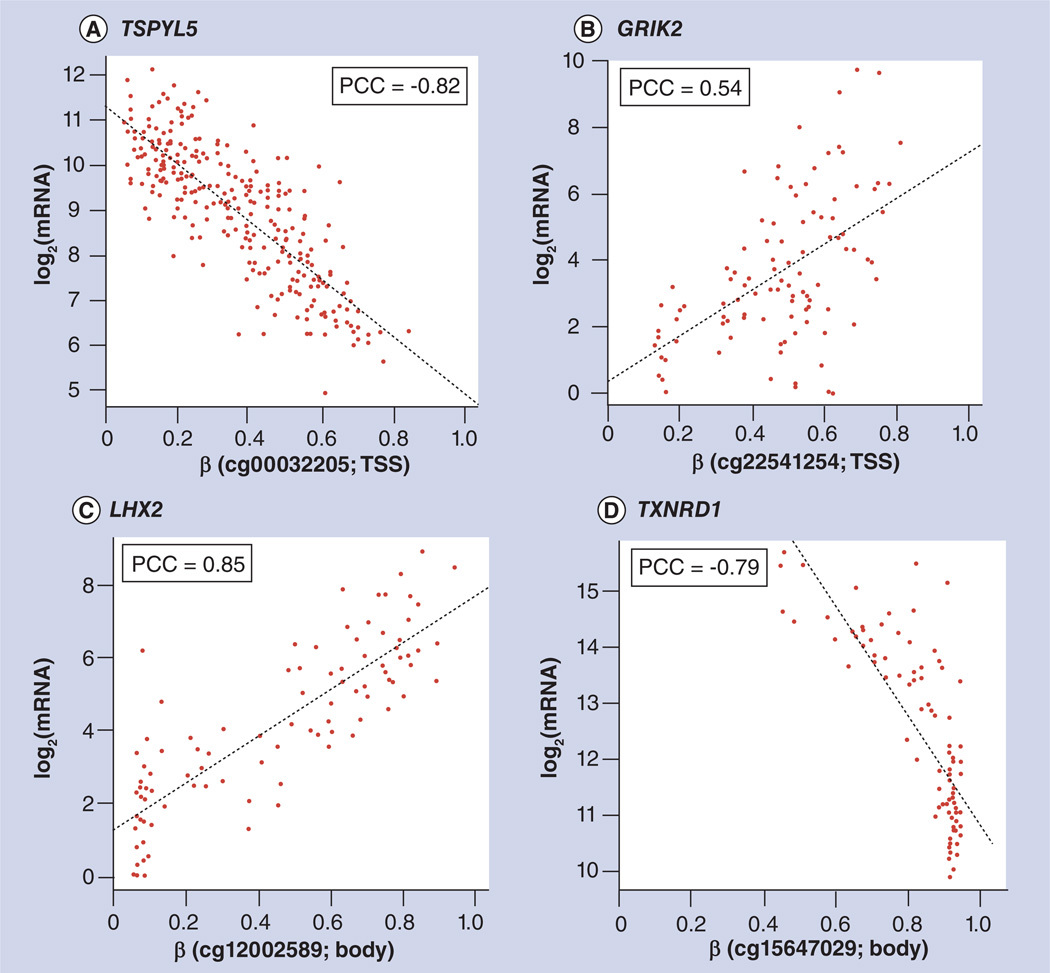
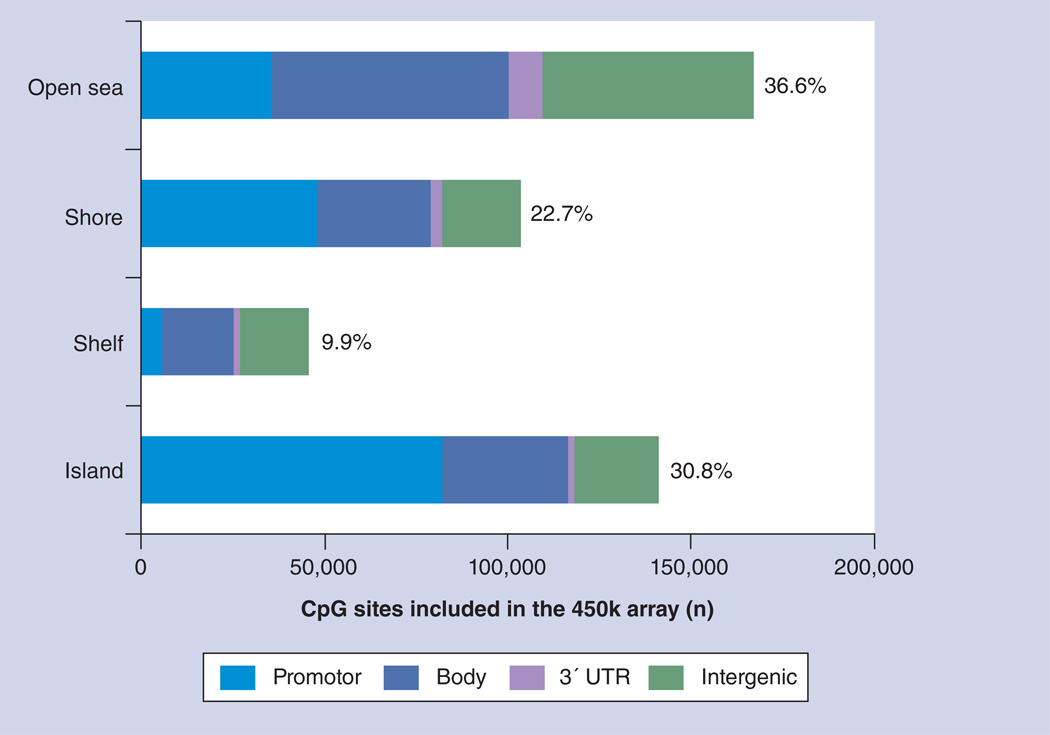
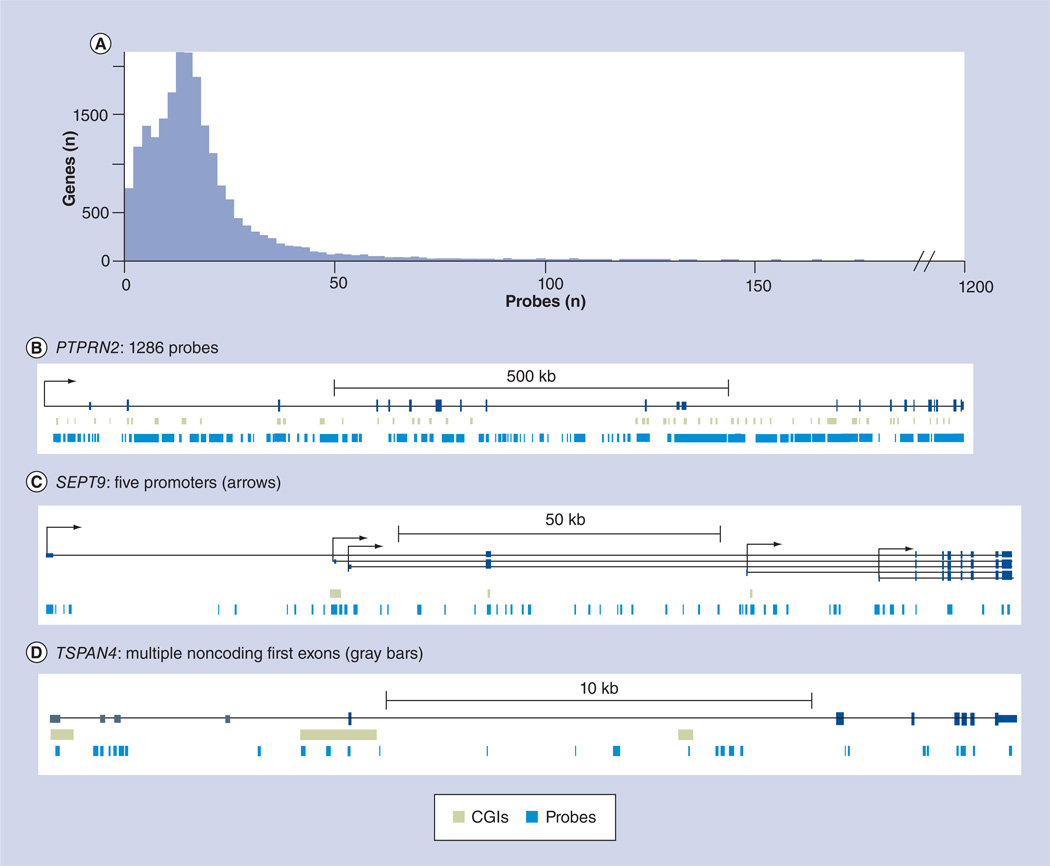
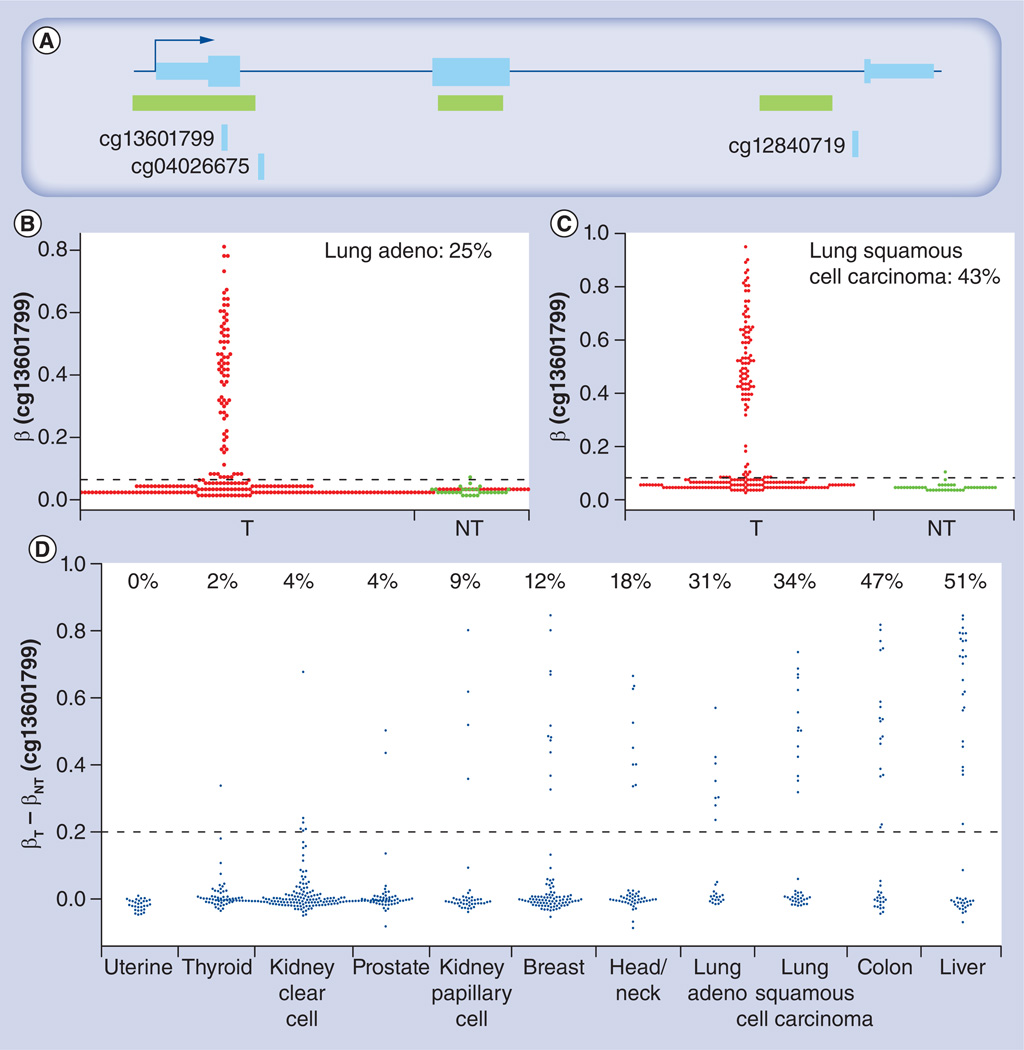
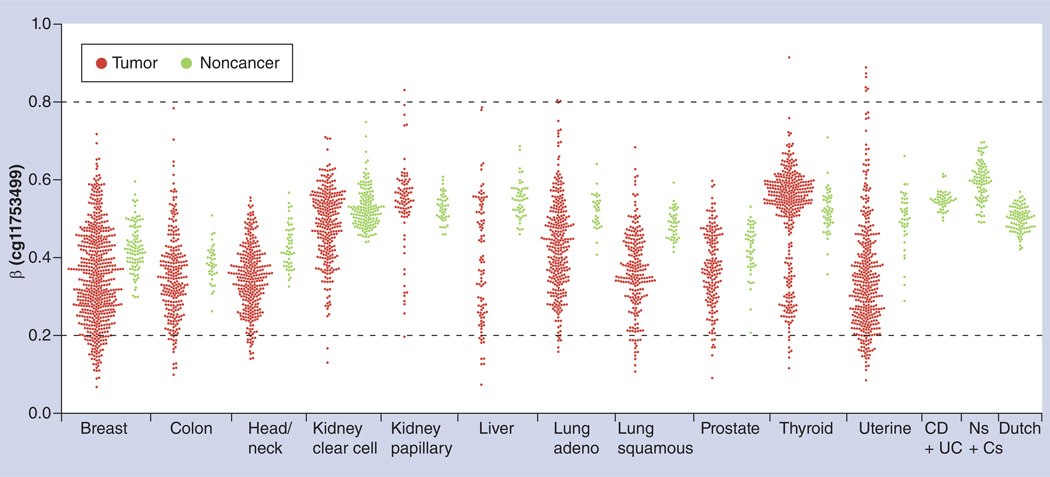
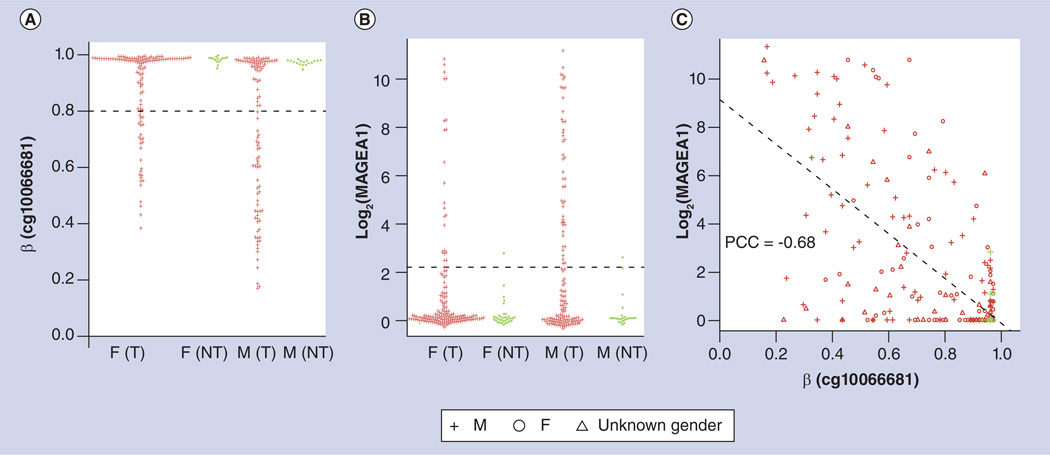
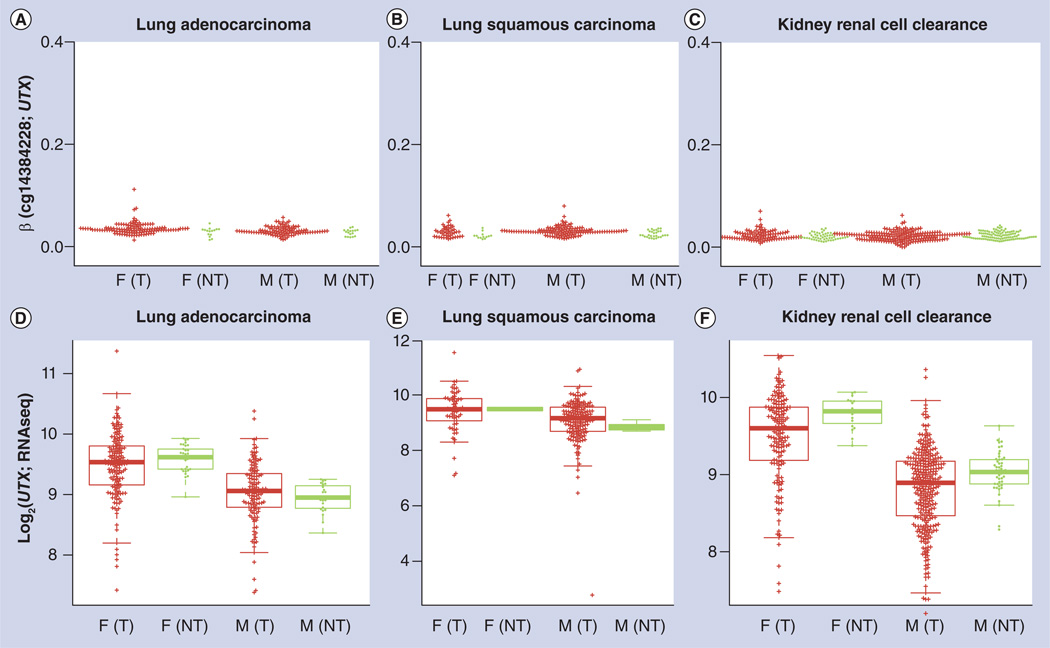
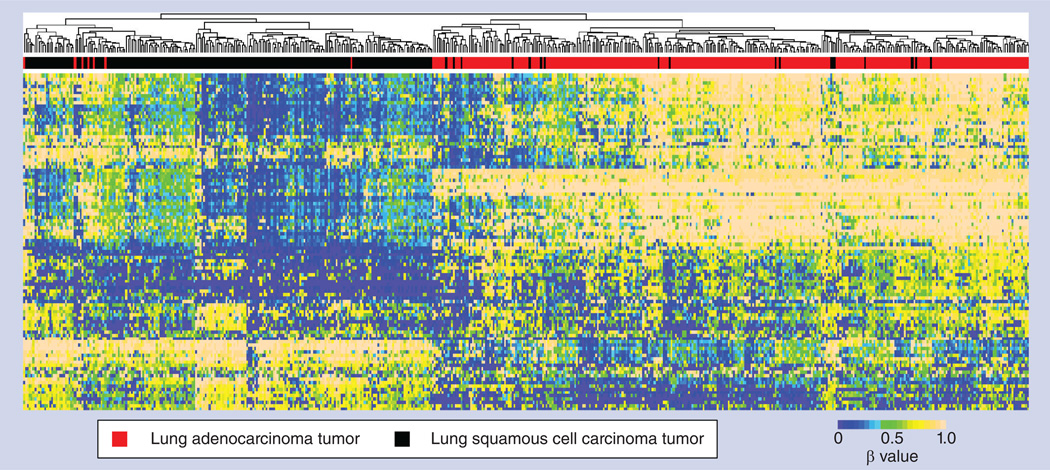
References
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–470. - PubMed
-
- Ren B, Robert F, Wyrick JJ, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290(5500):2306–2309. - PubMed
-
- Wang DG, Fan JB, Siao CJ, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280(5366):1077–1082. - PubMed
-
- Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295. - PubMed
-
-
Laird PW. Principles challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010;11(3):191–203.▪▪ Excellent review of DNA methylation measurements and analysis.
-
Websites
-
- Nature. Nature ENCODE explorer. www.nature.com/encode.
-
- ENCODE Data Coordination Center at UCSC. ENCODE common cell types. http://genome.ucsc.edu/ENCODE/cellTypes.html.
-
- NIH Roadmap Epigenomics Project. www.roadmapepigenomics.org.
-
- NIH working definition of bioinformatics and computational biology. www.bisti.nih.gov/docs/CompuBioDef.pdf.
-
- National Center for Biotechnology Information. RefSeq: NCBI Reference Sequence Database. www.ncbi.nlm.nih.gov/RefSeq/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources